We had a busy Spring, with three students successfully defending their honors theses… congrats to Adam, Brian, and Cathy!
THE CAO LAB
Organic materials research at a primarily undergraduate institution
NEWS
More azaacene diimides
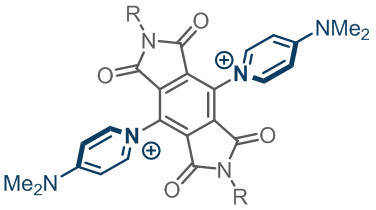
Our diimides made their way to Germany and the Bunz Lab and turned into a fun series of azaacenes with NIR absorptions and great electron affinities. Read more here.
Conor defends Honors thesis
Conor fended off the pandemic with a presentation and successful defense of his Honors work. Conor will stick around Macalester for a little, take a gap year, then pursue a PhD at UC Berkeley. Congrats Conor!
Dennis Receives Tenure
It’s official! Dennis has received tenure at Macalester, which was only possible because of the contributions of everyone who spent time in the Cao Lab. We’re looking forward to the next ??? years!
RESEARCH

PEOPLE

Summer 2019
From Left to Right: Dennis, Conor, Grace, Kellie, Stella

Summer 2018
From Left to Right: Dennis, Minji, Jay, Steven, Kellie, Stella

Summer 2017
From Left to Right: Fernando, Dennis, Nhu, Stella, Eman, Kellie, Hiywot, Mohammed, Qifan

Summer 2016
From Left to Right: top, Qifan, Mohammed, Joe, Andrew; bottom, Eman, Kofi, Penny, Dennis
Dennis Cao
Dennis was born in Konstanz, Germany and grew up in San Diego, California. He began his education at UC Berkeley in 2006 and did his undergrad research with Dr. Yi Liu at Lawrence Berkeley National Laboratory. In 2010, Dennis found his way into grad school and Prof. J. Fraser Stoddart's lab at Northwestern where he crystallized metal-organic frameworks and organic ferroelectrics before graduating in June 2014. He then did a postdoctoral stint in Prof. Samuel I. Stupp’s group, working on self-assembling materials. Dennis' independent career at Macalester College began in August 2015, where he is teaching organic chemistry and mentoring research students. Outside of chemistry, Dennis tries to balance his lifestyle with passions for photography, travel, Houston Rockets basketball, and various random hobbies.

PUBLICATIONS
2024
Gebresilassie, Feven Leake; Kim, Min Ji; Castellanos, Daniela; Broderick, Conor H.; Ngo, Steven M.; Jr., Victor G. Young; Cao, Dennis D.
Bisphosphonium Benzene Diimides Journal Article
In: Chemistry – A European Journal, pp. e202402791, 2024.
@article{https://doi.org/10.1002/chem.202402791,
title = {Bisphosphonium Benzene Diimides},
author = {Feven Leake Gebresilassie and Min Ji Kim and Daniela Castellanos and Conor H. Broderick and Steven M. Ngo and Victor G. Young Jr. and Dennis D. Cao},
url = {https://chemistry-europe.onlinelibrary.wiley.com/doi/abs/10.1002/chem.202402791},
doi = {https://doi.org/10.1002/chem.202402791},
year = {2024},
date = {2024-07-30},
journal = {Chemistry – A European Journal},
pages = {e202402791},
abstract = {Abstract The incorporation of cationic groups onto electron-poor compounds is a viable strategy for achieving potent electron acceptors, as evidenced by reports of air-stable radical forms of large aromatic diimides such as naphthalene and perylene diimides. These ions have also been observed to exhibit anion–π interaction tendencies of interest in molecular recognition applications. The benefits of phosphonium incorporation, however, have not yet been extended to the smallest benzene diimides. Here, we report that dibrominated pyromellitic diimide and mellophanic diimide both readily undergo substitution reactions with phosphine sources to yield bisphosphonium compounds. In the single crystalline form, these dications display anion-π interactions and, in the case of mellophanic diimide, the stabilization of a bromide–water H−bonding ring pattern. The reaction of these dications with chemical reductants readily provides the singly and doubly reduced redox states, which were characterized by UV-vis spectroscopy and found to exhibit intense absorptions extending into the near-IR region. Taken together, this work demonstrates that phosphonium incorporation onto congested aromatic diimide scaffolds is synthetically viable and produces unusual electron-poor compounds.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Abstract The incorporation of cationic groups onto electron-poor compounds is a viable strategy for achieving potent electron acceptors, as evidenced by reports of air-stable radical forms of large aromatic diimides such as naphthalene and perylene diimides. These ions have also been observed to exhibit anion–π interaction tendencies of interest in molecular recognition applications. The benefits of phosphonium incorporation, however, have not yet been extended to the smallest benzene diimides. Here, we report that dibrominated pyromellitic diimide and mellophanic diimide both readily undergo substitution reactions with phosphine sources to yield bisphosphonium compounds. In the single crystalline form, these dications display anion-π interactions and, in the case of mellophanic diimide, the stabilization of a bromide–water H−bonding ring pattern. The reaction of these dications with chemical reductants readily provides the singly and doubly reduced redox states, which were characterized by UV-vis spectroscopy and found to exhibit intense absorptions extending into the near-IR region. Taken together, this work demonstrates that phosphonium incorporation onto congested aromatic diimide scaffolds is synthetically viable and produces unusual electron-poor compounds.
Bass, Adam D; Castellanos, Daniela; Calicdan, Xavier A; Cao, Dennis D
Synthesis and characterization of 1,2,3,4-naphthalene and anthracene diimides Journal Article
In: Beilstein Journal of Organic Chemistry, vol. 20, pp. 1767–1772, 2024, ISSN: 1860-5397.
@article{bassSynthesisCharacterization4naphthalene2024,
title = {Synthesis and characterization of 1,2,3,4-naphthalene and anthracene diimides},
author = {Adam D Bass and Daniela Castellanos and Xavier A Calicdan and Dennis D Cao},
url = {https://www.beilstein-journals.org/bjoc/articles/20/155},
doi = {10.3762/bjoc.20.155},
issn = {1860-5397},
year = {2024},
date = {2024-07-01},
urldate = {2024-08-07},
journal = {Beilstein Journal of Organic Chemistry},
volume = {20},
pages = {1767–1772},
abstract = {We report the synthesis and characterization of naphthalene and anthracene scaffolds end-capped by cyclic imides. The solid-state structures of the N-phenyl derivatives, determined by X-ray crystallography, reveal changes in packing preference based on the number of aromatic rings in the core. The optical and electronic properties of the title compounds compare favorably with other previously described isomers and expand the toolbox of electron-deficient aromatic compounds available to organic materials chemists.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
We report the synthesis and characterization of naphthalene and anthracene scaffolds end-capped by cyclic imides. The solid-state structures of the N-phenyl derivatives, determined by X-ray crystallography, reveal changes in packing preference based on the number of aromatic rings in the core. The optical and electronic properties of the title compounds compare favorably with other previously described isomers and expand the toolbox of electron-deficient aromatic compounds available to organic materials chemists.
2023
Hippchen, Nikolai; Heinzel, Elena; Zhang, Cathy K.; Jäger, Patrick; Elter, Maximilian; Ludwig, Philipp; Rominger, Frank; Freudenberg, Jan; Cao, Dennis D.; Bunz, Uwe H. F.
Second Generation of Cata-Annulated Azaacene Bisimides: Towards Electron-Accepting Materials Journal Article
In: ChemPlusChem, vol. 88, no. 5, pp. e202300158, 2023, ISSN: 2192-6506, (_eprint: https://onlinelibrary.wiley.com/doi/pdf/10.1002/cplu.202300158).
@article{hippchenSecondGenerationCataAnnulated2023,
title = {Second Generation of Cata-Annulated Azaacene Bisimides: Towards Electron-Accepting Materials},
author = {Nikolai Hippchen and Elena Heinzel and Cathy K. Zhang and Patrick Jäger and Maximilian Elter and Philipp Ludwig and Frank Rominger and Jan Freudenberg and Dennis D. Cao and Uwe H. F. Bunz},
url = {https://onlinelibrary.wiley.com/doi/abs/10.1002/cplu.202300158},
doi = {10.1002/cplu.202300158},
issn = {2192-6506},
year = {2023},
date = {2023-01-01},
urldate = {2023-07-21},
journal = {ChemPlusChem},
volume = {88},
number = {5},
pages = {e202300158},
abstract = {This work presents the 2nd generation of cata-annulated azaacene bisimides with increased electron affinities (up to −4.38 eV) compared to their consaguine conventional azaacenes. These compounds were synthesized via Buchwald–Hartwig coupling followed by oxidation with MnO2. Crystal structure engineering through variation of the bisimide substituents furnished crystalline derivatives suitable for proof of concept organic field effect transistors with electron mobilities up to 2.2×10−4 cm2(Vs)−1. Moreover, we were able to characterize the charge carrying species, the radical anion, using electron paramagnetic resonance and absorption measurements.},
note = {_eprint: https://onlinelibrary.wiley.com/doi/pdf/10.1002/cplu.202300158},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
This work presents the 2nd generation of cata-annulated azaacene bisimides with increased electron affinities (up to −4.38 eV) compared to their consaguine conventional azaacenes. These compounds were synthesized via Buchwald–Hartwig coupling followed by oxidation with MnO2. Crystal structure engineering through variation of the bisimide substituents furnished crystalline derivatives suitable for proof of concept organic field effect transistors with electron mobilities up to 2.2×10−4 cm2(Vs)−1. Moreover, we were able to characterize the charge carrying species, the radical anion, using electron paramagnetic resonance and absorption measurements.
Cao, Dennis D.
Self-assembly Templated by Radical–Radical Interactions Book Section
In: Supramolecular Nanotechnology, pp. 231–243, John Wiley & Sons, Ltd, 2023, ISBN: 978-3-527-83404-4, (Section: 7 _eprint: https://onlinelibrary.wiley.com/doi/pdf/10.1002/9783527834044.ch7).
@incollection{caoSelfassemblyTemplatedRadical2023,
title = {Self-assembly Templated by Radical–Radical Interactions},
author = {Dennis D. Cao},
url = {https://onlinelibrary.wiley.com/doi/abs/10.1002/9783527834044.ch7},
doi = {10.1002/9783527834044.ch7},
isbn = {978-3-527-83404-4},
year = {2023},
date = {2023-01-01},
urldate = {2023-07-22},
booktitle = {Supramolecular Nanotechnology},
pages = {231–243},
publisher = {John Wiley & Sons, Ltd},
abstract = {This chapter is intended to provide a tutorial-type overview of radical-mediated self-assembly. It begins with a historical introduction to organic radicals and a selection of experimental findings that foreshadowed the development of radical–radical interactions. Supramolecular and molecular examples for stabilizing these species are described subsequently, before a discussion of how these radical–radical interactions can be used to create larger architectures and assemblies.},
note = {Section: 7
_eprint: https://onlinelibrary.wiley.com/doi/pdf/10.1002/9783527834044.ch7},
keywords = {},
pubstate = {published},
tppubtype = {incollection}
}
This chapter is intended to provide a tutorial-type overview of radical-mediated self-assembly. It begins with a historical introduction to organic radicals and a selection of experimental findings that foreshadowed the development of radical–radical interactions. Supramolecular and molecular examples for stabilizing these species are described subsequently, before a discussion of how these radical–radical interactions can be used to create larger architectures and assemblies.
2022
Zou, Brian; Stellmach, Kellie A.; Luo, Stella M.; Gebresilassie, Feven L.; Jung, Healeam; Zhang, Cathy K.; Bass, Adam D.; Janzen, Daron E.; Cao, Dennis D.
Improved Syntheses of Halogenated Benzene-1,2,3,4-Tetracarboxylic Diimides Journal Article
In: The Journal of Organic Chemistry, vol. 87, pp. 13604–13615, 2022, ISSN: 0022-3263, 1520-6904.
@article{zouImprovedSynthesesHalogenated2022,
title = {Improved Syntheses of Halogenated Benzene-1,2,3,4-Tetracarboxylic Diimides},
author = {Brian Zou and Kellie A. Stellmach and Stella M. Luo and Feven L. Gebresilassie and Healeam Jung and Cathy K. Zhang and Adam D. Bass and Daron E. Janzen and Dennis D. Cao},
url = {https://pubs.acs.org/doi/10.1021/acs.joc.2c01241},
doi = {10.1021/acs.joc.2c01241},
issn = {0022-3263, 1520-6904},
year = {2022},
date = {2022-10-01},
urldate = {2022-11-03},
journal = {The Journal of Organic Chemistry},
volume = {87},
pages = {13604–13615},
abstract = {The preparation of halogenated benzene-1,2,3,4tetracarboxylic diimide derivatives is challenging because of the possibility of competitive incorrect cyclizations and SNAr reactivity. Here, we demonstrate that bypassing traditional cyclic anhydrides and instead directly reacting dihalobenzene-1,2,3,4-tetracarboxylic acids with primary amines in acetic acid solvent successfully provides a range of desirable ortho-diimide products in good yields. Furthermore, we demonstrate that sterically challenging Nderivatizations can be readily achieved under microwave reactor conditions. The halogenated diimides described here are attractive building blocks for organic materials chemistry.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The preparation of halogenated benzene-1,2,3,4tetracarboxylic diimide derivatives is challenging because of the possibility of competitive incorrect cyclizations and SNAr reactivity. Here, we demonstrate that bypassing traditional cyclic anhydrides and instead directly reacting dihalobenzene-1,2,3,4-tetracarboxylic acids with primary amines in acetic acid solvent successfully provides a range of desirable ortho-diimide products in good yields. Furthermore, we demonstrate that sterically challenging Nderivatizations can be readily achieved under microwave reactor conditions. The halogenated diimides described here are attractive building blocks for organic materials chemistry.
Cao, Dennis D.
RealOrganicChemistry.org: A Collection of Introductory-Appropriate Organic Chemistry Reactions Journal Article
In: Journal of Chemical Education, vol. 99, no. 8, pp. 3049–3052, 2022, ISSN: 0021-9584, 1938-1328.
@article{caoRealOrganicChemistryOrgCollection2022,
title = {RealOrganicChemistry.org: A Collection of Introductory-Appropriate Organic Chemistry Reactions},
author = {Dennis D. Cao},
url = {https://pubs.acs.org/doi/10.1021/acs.jchemed.2c00424},
doi = {10.1021/acs.jchemed.2c00424},
issn = {0021-9584, 1938-1328},
year = {2022},
date = {2022-08-01},
urldate = {2022-10-14},
journal = {Journal of Chemical Education},
volume = {99},
number = {8},
pages = {3049–3052},
abstract = {For most students, completing practice problems is an essential prerequisite for success in organic chemistry courses. Practice work, however, is sometimes perceived to be easier than or even not related to the types of chemistry that students see in assessments and “real-world” science. This negative view may contribute to a reduced level of engagement with course content and scientific learning practices. To combat this perception, a collection of introductory level-appropriate organic reactions bearing literature citations has been assembled and made available at RealOrganicChemistry.org. Portions of this collection were crowdsourced by introductory organic chemistry students through a three-part literature assignment intended for students reading the chemical literature for the first time. For students, RealOrganicChemistry.org is intended to provide a wealth of practice opportunities to complement conventional learning resources. For instructors, RealOrganicChemistry.org hosts a simple-to-complex range of reaction examples that are useful for literature-backed teaching efforts.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
For most students, completing practice problems is an essential prerequisite for success in organic chemistry courses. Practice work, however, is sometimes perceived to be easier than or even not related to the types of chemistry that students see in assessments and “real-world” science. This negative view may contribute to a reduced level of engagement with course content and scientific learning practices. To combat this perception, a collection of introductory level-appropriate organic reactions bearing literature citations has been assembled and made available at RealOrganicChemistry.org. Portions of this collection were crowdsourced by introductory organic chemistry students through a three-part literature assignment intended for students reading the chemical literature for the first time. For students, RealOrganicChemistry.org is intended to provide a wealth of practice opportunities to complement conventional learning resources. For instructors, RealOrganicChemistry.org hosts a simple-to-complex range of reaction examples that are useful for literature-backed teaching efforts.
2021
Elter, Maximilian; Ahrens, Lukas; Luo, Stella M.; Rominger, Frank; Freudenberg, Jan; Cao, Dennis D.; Bunz, Uwe H. F.
Cata-Annulated Azaacene Bisimides Journal Article
In: Chemistry – A European Journal, vol. 27, no. 48, pp. 12284-12288, 2021.
@article{https://doi.org/10.1002/chem.202101573,
title = {Cata-Annulated Azaacene Bisimides},
author = {Maximilian Elter and Lukas Ahrens and Stella M. Luo and Frank Rominger and Jan Freudenberg and Dennis D. Cao and Uwe H. F. Bunz},
url = {https://chemistry-europe.onlinelibrary.wiley.com/doi/abs/10.1002/chem.202101573},
doi = {https://doi.org/10.1002/chem.202101573},
year = {2021},
date = {2021-01-01},
journal = {Chemistry – A European Journal},
volume = {27},
number = {48},
pages = {12284-12288},
abstract = {Abstract Ultra-electron-deficient azaacenes were synthesized via Buchwald-Hartwig coupling of ortho-diaminoarenes with chlorinated mellophanic diimide followed by oxidation of the intermediate N,N’-dihydro compounds with MnO2 or PbO2. The resulting cata-annulated bisimide azaacenes have ultrahigh electron affinities with first reduction potentials as low as −0.35 V recorded for a tetraazapentacene. Attempts to prepare a tetrakis(dicarboximide)tetraazaheptacene resulted in the formation of a symmetric butterfly dimer.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Abstract Ultra-electron-deficient azaacenes were synthesized via Buchwald-Hartwig coupling of ortho-diaminoarenes with chlorinated mellophanic diimide followed by oxidation of the intermediate N,N’-dihydro compounds with MnO2 or PbO2. The resulting cata-annulated bisimide azaacenes have ultrahigh electron affinities with first reduction potentials as low as −0.35 V recorded for a tetraazapentacene. Attempts to prepare a tetrakis(dicarboximide)tetraazaheptacene resulted in the formation of a symmetric butterfly dimer.
2019
Kim, Min Ji; Luo, Stella M; Greenlee, Andrew J; Young, Victor G; Cao, Dennis D
A Highly Stabilized Phosphonium Ylide that Forms Supramolecular Dimers in Solution and the Solid State Journal Article
In: Chemistry – A European Journal, vol. 0, no. 0, 2019, ISSN: 1521-3765.
@article{kim_highly_nodate,
title = {A Highly Stabilized Phosphonium Ylide that Forms Supramolecular Dimers in Solution and the Solid State},
author = {Min Ji Kim and Stella M Luo and Andrew J Greenlee and Victor G Young and Dennis D Cao},
url = {https://onlinelibrary.wiley.com/doi/abs/10.1002/chem.201904131},
doi = {10.1002/chem.201904131},
issn = {1521-3765},
year = {2019},
date = {2019-10-26},
urldate = {2019-10-28},
journal = {Chemistry – A European Journal},
volume = {0},
number = {0},
abstract = {This work describes the unexpected formation of an unusual phosphonium ylide when attempting the synthesis of bisphosphonium pyromellitic diimides. Spectroscopic and crystallographic characterization reveals that a combination of π–π and CH⋅⋅⋅O interactions leads to supramolecular homodimerization of the ylide both in solution and in the solid-state. Only strong acids are able to protonate the ylide, which is otherwise inert to Wittig and alkylation reactivity. Taken together, these observations indicate that this compound is one of the most highly stabilized phosphonium ylides discovered to date.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
This work describes the unexpected formation of an unusual phosphonium ylide when attempting the synthesis of bisphosphonium pyromellitic diimides. Spectroscopic and crystallographic characterization reveals that a combination of π–π and CH⋅⋅⋅O interactions leads to supramolecular homodimerization of the ylide both in solution and in the solid-state. Only strong acids are able to protonate the ylide, which is otherwise inert to Wittig and alkylation reactivity. Taken together, these observations indicate that this compound is one of the most highly stabilized phosphonium ylides discovered to date.
Luo, Stella M; Stellmach, Kellie A; Ikuzwe, Stella M; Cao, Dennis D
Redox-Active Heteroacene Chromophores Derived from a Nonlinear Aromatic Diimide Journal Article
In: The Journal of Organic Chemistry, vol. 84, no. 16, pp. 10362–10370, 2019, ISSN: 0022-3263.
@article{luo_redox-active_2019,
title = {Redox-Active Heteroacene Chromophores Derived from a Nonlinear Aromatic Diimide},
author = {Stella M Luo and Kellie A Stellmach and Stella M Ikuzwe and Dennis D Cao},
url = {https://doi.org/10.1021/acs.joc.9b01502},
doi = {10.1021/acs.joc.9b01502},
issn = {0022-3263},
year = {2019},
date = {2019-01-01},
urldate = {2019-07-31},
journal = {The Journal of Organic Chemistry},
volume = {84},
number = {16},
pages = {10362--10370},
abstract = {This work describes a three-step chromatography-free protocol for the synthesis of a novel organic materials building block, dichlorinated mellophanic diimide (MDI), that is shown to undergo nucleophilic substitution with a variety of ortho disubstituted benzenes to yield a series of chromophores. Furthermore, 1,2,4,5-tetrasubstituted benzenes can be used to synthesize tetraimide heteropentacene derivatives endcapped by MDI motifs. The fine-tuning effects of heteroatom identity were investigated by UV–vis and fluorescence spectroscopy, cyclic and differential pulse voltammetries, and density functional theory calculations. Oxidation of diamino MDI derivatives yields di- and tetraimide functionalized azaacenes with significantly lowered LUMO levels (down to −4.49 eV), narrowed band gaps (down to 1.81 eV), and high molar absorptivities (up to 84,000 M–1 cm–1).},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
This work describes a three-step chromatography-free protocol for the synthesis of a novel organic materials building block, dichlorinated mellophanic diimide (MDI), that is shown to undergo nucleophilic substitution with a variety of ortho disubstituted benzenes to yield a series of chromophores. Furthermore, 1,2,4,5-tetrasubstituted benzenes can be used to synthesize tetraimide heteropentacene derivatives endcapped by MDI motifs. The fine-tuning effects of heteroatom identity were investigated by UV–vis and fluorescence spectroscopy, cyclic and differential pulse voltammetries, and density functional theory calculations. Oxidation of diamino MDI derivatives yields di- and tetraimide functionalized azaacenes with significantly lowered LUMO levels (down to −4.49 eV), narrowed band gaps (down to 1.81 eV), and high molar absorptivities (up to 84,000 M–1 cm–1).
2018
Kahn, Penelope C; Cao, Dennis D; Burns, Mercedes; Boyer, Sarah L
Nuptial gift chemistry reveals convergent evolution correlated with antagonism in mating systems of harvestmen (Arachnida, Opiliones) Journal Article
In: Ecology and Evolution, vol. 8, no. 14, pp. 7103-7110, 2018.
@article{doi:10.1002/ece3.4232,
title = {Nuptial gift chemistry reveals convergent evolution correlated with antagonism in mating systems of harvestmen (Arachnida, Opiliones)},
author = {Penelope C Kahn and Dennis D Cao and Mercedes Burns and Sarah L Boyer},
url = {https://onlinelibrary.wiley.com/doi/abs/10.1002/ece3.4232},
doi = {10.1002/ece3.4232},
year = {2018},
date = {2018-06-22},
journal = {Ecology and Evolution},
volume = {8},
number = {14},
pages = {7103-7110},
abstract = {Abstract Nuptial gifts are material donations given from male to female before or during copulation and are subject to sexual selection in a wide variety of taxa. The harvestman genus Leiobunum has emerged as a model system for understanding the evolution of reproductive morphology and behavior, as transitions between solicitous and antagonistic modes of courtship have occurred multiple times within the lineage and are correlated with convergence in genital morphology. We analyzed the free amino acid content of nuptial gift secretions from five species of Leiobunum using gas chromatography–mass spectrometry. Multivariate analysis of the free amino acid profiles revealed that, rather than clustering based on phylogenetic relationships, nuptial gift chemical composition was better predicted by genital morphology and behavior, suggesting that convergent evolution has acted on the chemical composition of the nuptial gift. In addition, we found that, species with solicitous courtship produce gifts consisting of a 19% larger proportion of essential amino acids as compared to those with more antagonistic courtship interactions. This work represents the first comparative study of nuptial gift chemistry within a phylogenetic framework in any animal group and as such contributes to our understanding of the evolution of reproductive diversity and the participant role of nuptial gift chemistry in mating system transitions.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Abstract Nuptial gifts are material donations given from male to female before or during copulation and are subject to sexual selection in a wide variety of taxa. The harvestman genus Leiobunum has emerged as a model system for understanding the evolution of reproductive morphology and behavior, as transitions between solicitous and antagonistic modes of courtship have occurred multiple times within the lineage and are correlated with convergence in genital morphology. We analyzed the free amino acid content of nuptial gift secretions from five species of Leiobunum using gas chromatography–mass spectrometry. Multivariate analysis of the free amino acid profiles revealed that, rather than clustering based on phylogenetic relationships, nuptial gift chemical composition was better predicted by genital morphology and behavior, suggesting that convergent evolution has acted on the chemical composition of the nuptial gift. In addition, we found that, species with solicitous courtship produce gifts consisting of a 19% larger proportion of essential amino acids as compared to those with more antagonistic courtship interactions. This work represents the first comparative study of nuptial gift chemistry within a phylogenetic framework in any animal group and as such contributes to our understanding of the evolution of reproductive diversity and the participant role of nuptial gift chemistry in mating system transitions.
Greenlee, Andrew J; Ofosu, Charles K; Xiao, Qifan; Modan, Mohammed M; Janzen, Daron E; Cao, Dennis D
Pyridinium-Functionalized Pyromellitic Diimides with Stabilized Radical Anion States Journal Article
In: ACS Omega, vol. 3, no. 1, pp. 240–245, 2018, ISSN: 2470-1343.
@article{greenlee_pyridinium-functionalized_2018,
title = {Pyridinium-Functionalized Pyromellitic Diimides with Stabilized Radical Anion States},
author = {Andrew J Greenlee and Charles K Ofosu and Qifan Xiao and Mohammed M Modan and Daron E Janzen and Dennis D Cao},
url = {http://pubs.acs.org/doi/abs/10.1021/acsomega.7b01887},
doi = {10.1021/acsomega.7b01887},
issn = {2470-1343},
year = {2018},
date = {2018-01-01},
urldate = {2018-01-09},
journal = {ACS Omega},
volume = {3},
number = {1},
pages = {240--245},
abstract = {In this work, we report the stabilization of the reduced states of pyromellitic diimide by charge-balancing the imide radical anions with cationic pyridinium groups attached to the aromatic core. This structural modification is confirmed by single-crystal X-ray diffraction analysis. Characterization by (spectro)electrochemical experiments and computations reveal that the addition of cationic groups to an already electron-deficient ring system results in up to +0.57 V shifts in reduction potentials, largely as a consequence of charge screening and lowest unoccupied molecular orbital-lowering effects. This formal charge-balancing approach to stabilizing the reduced states of electron-deficient pyromellitic diimides will facilitate their incorporation into spin-based optoelectronic materials and devices.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
In this work, we report the stabilization of the reduced states of pyromellitic diimide by charge-balancing the imide radical anions with cationic pyridinium groups attached to the aromatic core. This structural modification is confirmed by single-crystal X-ray diffraction analysis. Characterization by (spectro)electrochemical experiments and computations reveal that the addition of cationic groups to an already electron-deficient ring system results in up to +0.57 V shifts in reduction potentials, largely as a consequence of charge screening and lowest unoccupied molecular orbital-lowering effects. This formal charge-balancing approach to stabilizing the reduced states of electron-deficient pyromellitic diimides will facilitate their incorporation into spin-based optoelectronic materials and devices.
2017
Tayi, Alok S.; Shveyd, Alexander K.; Sue, Andrew C. -H.; Szarko, Jodi M.; Rolczynski, Brian S.; Cao, Dennis; Kennedy, T. Jackson; Sarjeant, Amy A.; Stern, Charlotte L.; Paxton, Walter F.; Wu, Wei; Dey, Sanjeev K.; Fahrenbach, Albert C.; Guest, Jeffrey R.; Mohseni, Hooman; Chen, Lin X.; Wang, Kang L.; Stoddart, J. Fraser; Stupp, Samuel I.
Tayi et al. reply to G. D’Avino et al. (regarding Nature 488, 485–489 (2012)) Journal Article
In: Nature, vol. 547, no. 7662, pp. E14–E15, 2017, ISSN: 0028-0836.
@article{tayi_tayi_2017,
title = {Tayi et al. reply to G. D’Avino et al. (regarding Nature 488, 485–489 (2012))},
author = {Tayi, Alok S. and Shveyd, Alexander K. and Sue, Andrew C.-H. and Szarko, Jodi M. and Rolczynski, Brian S. and Cao, Dennis and Kennedy, T. Jackson and Sarjeant, Amy A. and Stern, Charlotte L. and Paxton, Walter F. and Wu, Wei and Dey, Sanjeev K. and Fahrenbach, Albert C. and Guest, Jeffrey R. and Mohseni, Hooman and Chen, Lin X. and Wang, Kang L. and Stoddart, J. Fraser and Stupp, Samuel I.},
url = {http://www.nature.com/nature/journal/v547/n7662/full/nature22802.html?WT.feed_name=subjects_physics},
doi = {10.1038/nature22802},
issn = {0028-0836},
year = {2017},
date = {2017-07-01},
urldate = {2017-07-13},
journal = {Nature},
volume = {547},
number = {7662},
pages = {E14--E15},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Narayanan, Ashwin; Cao, Dennis; Frazer, Laszlo; Tayi, Alok S.; Blackburn, Anthea K.; Sue, Andrew C. -H.; Ketterson, John B.; Stoddart, J. Fraser; Stupp, Samuel I.
Ferroelectric Polarization and Second Harmonic Generation in Supramolecular Cocrystals with Two Axes of Charge-Transfer Journal Article
In: Journal of the American Chemical Society, vol. 139, no. 27, pp. 9186–9191, 2017, ISSN: 0002-7863.
@article{narayanan_ferroelectric_2017,
title = {Ferroelectric Polarization and Second Harmonic Generation in Supramolecular Cocrystals with Two Axes of Charge-Transfer},
author = {Narayanan, Ashwin and Cao, Dennis and Frazer, Laszlo and Tayi, Alok S. and Blackburn, Anthea K. and Sue, Andrew C.-H. and Ketterson, John B. and Stoddart, J. Fraser and Stupp, Samuel I.},
url = {http://dx.doi.org/10.1021/jacs.7b02279},
doi = {10.1021/jacs.7b02279},
issn = {0002-7863},
year = {2017},
date = {2017-01-01},
journal = {Journal of the American Chemical Society},
volume = {139},
number = {27},
pages = {9186--9191},
abstract = {Ferroelectricity in organic materials remains a subject of great interest, given its potential impact as lightweight information storage media. Here we report supramolecular charge-transfer cocrystals formed by electron acceptor and donor molecules that exhibit ferroelectric behavior along two distinct crystallographic axes. The solid-state superstructure of the cocrystals reveals that a 2:1 ratio of acceptor to donor molecules assemble into nearly orthogonal mixed stacks in which the molecules are positioned for charge-transfer in face-to-face and edge-to-face orientations, held together by an extended hydrogen-bonding network. Polarization hysteresis was observed along the face-to-face and edge-to-face axes at room temperature. The noncentrosymmetric nature of the cocrystals, required to observe ferroelectric behavior, is demonstrated using second harmonic generation measurements. This finding suggests the possibility of designing supramolecular arrays in which organic molecules support multidimensional information storage.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Ferroelectricity in organic materials remains a subject of great interest, given its potential impact as lightweight information storage media. Here we report supramolecular charge-transfer cocrystals formed by electron acceptor and donor molecules that exhibit ferroelectric behavior along two distinct crystallographic axes. The solid-state superstructure of the cocrystals reveals that a 2:1 ratio of acceptor to donor molecules assemble into nearly orthogonal mixed stacks in which the molecules are positioned for charge-transfer in face-to-face and edge-to-face orientations, held together by an extended hydrogen-bonding network. Polarization hysteresis was observed along the face-to-face and edge-to-face axes at room temperature. The noncentrosymmetric nature of the cocrystals, required to observe ferroelectric behavior, is demonstrated using second harmonic generation measurements. This finding suggests the possibility of designing supramolecular arrays in which organic molecules support multidimensional information storage.
Wu, Yilei; Han, Ji-Min; Hong, Michael; Krzyaniak, Matthew D; Blackburn, Anthea K; Fernando, Isurika R; Cao, Dennis D; Wasielewski, Michael R; Stoddart, Fraser J
X-Shaped Oligomeric Pyromellitimide Polyradicals Journal Article
In: Journal of the American Chemical Society, 2017, ISSN: 0002-7863.
@article{wu_x-shaped_2017,
title = {X-Shaped Oligomeric Pyromellitimide Polyradicals},
author = {Yilei Wu and Ji-Min Han and Michael Hong and Matthew D Krzyaniak and Anthea K Blackburn and Isurika R Fernando and Dennis D Cao and Michael R Wasielewski and Fraser J Stoddart},
url = {http://dx.doi.org/10.1021/jacs.7b12124},
doi = {10.1021/jacs.7b12124},
issn = {0002-7863},
year = {2017},
date = {2017-01-01},
urldate = {2018-01-09},
journal = {Journal of the American Chemical Society},
abstract = {The synthesis of stable organic polyradicals is important for the development of magnetic materials. Herein we report the synthesis, isolation, and characterization of a series of X-shaped pyromellitimide (PI) oligomers (Xn-R},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The synthesis of stable organic polyradicals is important for the development of magnetic materials. Herein we report the synthesis, isolation, and characterization of a series of X-shaped pyromellitimide (PI) oligomers (Xn-R
2015
Sun, Junling; Wu, Yilei; Liu, Zhichang; Cao, Dennis; Wang, Yuping; Cheng, Chuyang; Chen, Dongyang; Wasielewski, Michael R.; Stoddart, J. Fraser
Visible Light-Driven Artificial Molecular Switch Actuated by Radical–Radical and Donor–Acceptor Interactions Journal Article
In: J. Phys. Chem. A, vol. 119, no. 24, pp. 6317–6325, 2015, ISSN: 1089-5639.
@article{sun_visible_2015,
title = {Visible Light-Driven Artificial Molecular Switch Actuated by Radical–Radical and Donor–Acceptor Interactions},
author = {Sun, Junling and Wu, Yilei and Liu, Zhichang and Cao, Dennis and Wang, Yuping and Cheng, Chuyang and Chen, Dongyang and Wasielewski, Michael R. and Stoddart, J. Fraser},
url = {http://dx.doi.org/10.1021/acs.jpca.5b04570},
doi = {10.1021/acs.jpca.5b04570},
issn = {1089-5639},
year = {2015},
date = {2015-06-01},
urldate = {2015-08-11},
journal = {J. Phys. Chem. A},
volume = {119},
number = {24},
pages = {6317--6325},
abstract = {We describe a visible light-driven switchable [2]catenane, composed of a Ru(bpy)32+ tethered cyclobis(paraquat-p-phenylene) (CBPQT4+) ring that is interlocked mechanically with a macrocyclic polyether consisting of electron-rich 1,5-dioxynaphthalene (DNP) and electron-deficient 4,4?-bipyridinium (BIPY2+) units. In the oxidized state, the CBPQT4+ ring encircles the DNP recognition site as a consequence of favorable donor?acceptor interactions. In the presence of an excess of triethanolamine (TEOA), visible light irradiation reduces the BIPY2+ units to BIPY(?+) radical cations under the influence of the photosensitizer Ru(bpy)32+, resulting in the movement of the CBPQT2(?+) ring from the DNP to the BIPY(?+) recognition site as a consequence of the formation of the more energetically favorable trisradical complex, BIPY(?+) ? CBPQT2(?+). Upon introducing O2 in the dark, the BIPY(?+) radical cations are oxidized back to BIPY2+ dications, leading to the reinstatement of the CBPQT4+ ring encircled around the DNP recognition site. Employing this strategy of redox control, we have demonstrated a prototypical molecular switch that can be manipulated photochemically and chemically by sequential reduction and oxidation.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
We describe a visible light-driven switchable [2]catenane, composed of a Ru(bpy)32+ tethered cyclobis(paraquat-p-phenylene) (CBPQT4+) ring that is interlocked mechanically with a macrocyclic polyether consisting of electron-rich 1,5-dioxynaphthalene (DNP) and electron-deficient 4,4?-bipyridinium (BIPY2+) units. In the oxidized state, the CBPQT4+ ring encircles the DNP recognition site as a consequence of favorable donor?acceptor interactions. In the presence of an excess of triethanolamine (TEOA), visible light irradiation reduces the BIPY2+ units to BIPY(?+) radical cations under the influence of the photosensitizer Ru(bpy)32+, resulting in the movement of the CBPQT2(?+) ring from the DNP to the BIPY(?+) recognition site as a consequence of the formation of the more energetically favorable trisradical complex, BIPY(?+) ? CBPQT2(?+). Upon introducing O2 in the dark, the BIPY(?+) radical cations are oxidized back to BIPY2+ dications, leading to the reinstatement of the CBPQT4+ ring encircled around the DNP recognition site. Employing this strategy of redox control, we have demonstrated a prototypical molecular switch that can be manipulated photochemically and chemically by sequential reduction and oxidation.
Sue, Andrew C. -H.; Mannige, Ranjan V.; Deng, Hexiang; Cao, Dennis; Wang, Cheng; Gándara, Felipe; Stoddart, J. Fraser; Whitelam, Stephen; Yaghi, Omar M.
Heterogeneity of Functional Groups in a Metal–organic Framework Displays Magic Number Ratios Journal Article
In: Proc. Nat. Acad. Sci., vol. 112, no. 18, pp. 5591–5596, 2015, ISSN: 0027-8424, 1091-6490.
@article{sue_heterogeneity_2015,
title = {Heterogeneity of Functional Groups in a Metal–organic Framework Displays Magic Number Ratios},
author = {Sue, Andrew C.-H. and Mannige, Ranjan V. and Deng, Hexiang and Cao, Dennis and Wang, Cheng and Gándara, Felipe and Stoddart, J. Fraser and Whitelam, Stephen and Yaghi, Omar M.},
url = {http://www.pnas.org/content/112/18/5591},
doi = {10.1073/pnas.1416417112},
issn = {0027-8424, 1091-6490},
year = {2015},
date = {2015-05-01},
urldate = {2015-05-20},
journal = {Proc. Nat. Acad. Sci.},
volume = {112},
number = {18},
pages = {5591--5596},
abstract = {Multiple organic functionalities can now be apportioned into nanoscale domains within a metal-coordinated framework, posing the following question: how do we control the resulting combination of “heterogeneity and order”? Here, we report the creation of a metal–organic framework, MOF-2000, whose two component types are incorporated in a 2:1 ratio, even when the ratio of component types in the starting solution is varied by an order of magnitude. Statistical mechanical modeling suggests that this robust 2:1 ratio has a nonequilibrium origin, resulting from kinetic trapping of component types during framework growth. Our simulations show how other “magic number” ratios of components can be obtained by modulating the topology of a framework and the noncovalent interactions between component types, a finding that may aid the rational design of functional multicomponent materials.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Multiple organic functionalities can now be apportioned into nanoscale domains within a metal-coordinated framework, posing the following question: how do we control the resulting combination of “heterogeneity and order”? Here, we report the creation of a metal–organic framework, MOF-2000, whose two component types are incorporated in a 2:1 ratio, even when the ratio of component types in the starting solution is varied by an order of magnitude. Statistical mechanical modeling suggests that this robust 2:1 ratio has a nonequilibrium origin, resulting from kinetic trapping of component types during framework growth. Our simulations show how other “magic number” ratios of components can be obtained by modulating the topology of a framework and the noncovalent interactions between component types, a finding that may aid the rational design of functional multicomponent materials.
2014
Liu, Zhichang; Liu, Guoliang; Wu, Yilei; Cao, Dennis; Sun, Junling; Schneebeli, Severin T.; Nassar, Majed S.; Mirkin, Chad A.; Stoddart, J. Fraser
Assembly of Supramolecular Nanotubes from Molecular Triangles and 1,2-Dihalohydrocarbons Journal Article
In: J. Am. Chem. Soc., vol. 136, no. 47, pp. 16651−16660, 2014, ISSN: 0002-7863.
@article{liu_assembly_2014,
title = {Assembly of Supramolecular Nanotubes from Molecular Triangles and 1,2-Dihalohydrocarbons},
author = {Liu, Zhichang and Liu, Guoliang and Wu, Yilei and Cao, Dennis and Sun, Junling and Schneebeli, Severin T. and Nassar, Majed S. and Mirkin, Chad A. and Stoddart, J. Fraser},
url = {http://dx.doi.org/10.1021/ja509480u},
doi = {10.1021/ja509480u},
issn = {0002-7863},
year = {2014},
date = {2014-11-01},
urldate = {2014-12-02},
journal = {J. Am. Chem. Soc.},
volume = {136},
number = {47},
pages = {16651−16660},
abstract = {Precise control of molecular assembly is a challenging goal facing supramolecular chemists. Herein, we report the highly specific assembly of a range of supramolecular nanotubes from the enantiomeric triangular naphthalenediimide-based macrocycles (RRRRRR)- and (SSSSSS)-NDI-? and a class of similar solvents, namely, the 1,2-dihalo-ethanes and -ethenes (DXEs). Three kinds of supramolecular nanotubes are formed from the columnar stacking of NDI-? units with a 60° mutual rotation angle as a result of cooperative [C?H···O] interactions, directing interactions of the [X···X]-bonded DXE chains inside the nanotubes and lateral [X···π] or [π···π] interactions. They include (i) semiflexible infinite nanotubes formed in the gel state from NDI-? and (E)-1,2-dichloroethene, (ii) rigid infinite nonhelical nanotubes produced in the solid state from NDI-? and BrCH2CH2Br, ClCH2CH2Br, and ClCH2CH2I, and (iii) a pair of rigid tetrameric, enantiomeric single-handed (P)- and (M)-helical nanotubes formed in the solid state from the corresponding (RRRRRR)- and (SSSSSS)-NDI-? with ClCH2CH2Cl. In case (i), only the electron-rich C═C double bond of (E)-1,2-dichloroethene facilitates the gelation of NDI-?. In cases (ii) and (iii), the lengths of anti-DXEs determine the translation of the chirality of NDI-? into the helicity of nanotubes. Only ClCH2CH2Cl induces single-handed helicity into the nanotubes. The subtle interplay of noncovalent bonding interactions, resulting from the tiny structural variations involving the DXE guests, is responsible for the diverse and highly specific assembly of NDI-?. This research highlights the critical role that guests play in constructing assembled superstructures of hosts and offers a novel approach to creating supramolecular nanotubes.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Precise control of molecular assembly is a challenging goal facing supramolecular chemists. Herein, we report the highly specific assembly of a range of supramolecular nanotubes from the enantiomeric triangular naphthalenediimide-based macrocycles (RRRRRR)- and (SSSSSS)-NDI-? and a class of similar solvents, namely, the 1,2-dihalo-ethanes and -ethenes (DXEs). Three kinds of supramolecular nanotubes are formed from the columnar stacking of NDI-? units with a 60° mutual rotation angle as a result of cooperative [C?H···O] interactions, directing interactions of the [X···X]-bonded DXE chains inside the nanotubes and lateral [X···π] or [π···π] interactions. They include (i) semiflexible infinite nanotubes formed in the gel state from NDI-? and (E)-1,2-dichloroethene, (ii) rigid infinite nonhelical nanotubes produced in the solid state from NDI-? and BrCH2CH2Br, ClCH2CH2Br, and ClCH2CH2I, and (iii) a pair of rigid tetrameric, enantiomeric single-handed (P)- and (M)-helical nanotubes formed in the solid state from the corresponding (RRRRRR)- and (SSSSSS)-NDI-? with ClCH2CH2Cl. In case (i), only the electron-rich C═C double bond of (E)-1,2-dichloroethene facilitates the gelation of NDI-?. In cases (ii) and (iii), the lengths of anti-DXEs determine the translation of the chirality of NDI-? into the helicity of nanotubes. Only ClCH2CH2Cl induces single-handed helicity into the nanotubes. The subtle interplay of noncovalent bonding interactions, resulting from the tiny structural variations involving the DXE guests, is responsible for the diverse and highly specific assembly of NDI-?. This research highlights the critical role that guests play in constructing assembled superstructures of hosts and offers a novel approach to creating supramolecular nanotubes.
Blackburn, Anthea K.; Sue, Andrew C. -H.; Shveyd, Alexander K.; Cao, Dennis; Tayi, Alok; Narayanan, Ashwin; Rolczynski, Brian S.; Szarko, Jodi M.; Bozdemir, Ozgur A.; Wakabayashi, Rie; Lehrman, Jessica A.; Kahr, Bart; Chen, Lin X.; Nassar, Majed S.; Stupp, Samuel I.; Stoddart, J. Fraser
Lock-Arm Supramolecular Ordering: A Molecular Construction Set for Cocrystallizing Organic Charge Transfer Complexes Journal Article
In: J. Am. Chem. Soc., vol. 136, pp. 17224−17235, 2014, ISSN: 0002-7863.
@article{blackburn_lock-arm_2014,
title = {Lock-Arm Supramolecular Ordering: A Molecular Construction Set for Cocrystallizing Organic Charge Transfer Complexes},
author = {Blackburn, Anthea K. and Sue, Andrew C.-H. and Shveyd, Alexander K. and Cao, Dennis and Tayi, Alok and Narayanan, Ashwin and Rolczynski, Brian S. and Szarko, Jodi M. and Bozdemir, Ozgur A. and Wakabayashi, Rie and Lehrman, Jessica A. and Kahr, Bart and Chen, Lin X. and Nassar, Majed S. and Stupp, Samuel I. and Stoddart, J. Fraser},
url = {http://dx.doi.org/10.1021/ja509442t},
doi = {10.1021/ja509442t},
issn = {0002-7863},
year = {2014},
date = {2014-11-01},
urldate = {2014-12-02},
journal = {J. Am. Chem. Soc.},
volume = {136},
pages = {17224−17235},
abstract = {Organic charge transfer cocrystals are inexpensive, modular, and solution-processable materials that are able, in some instances, to exhibit properties such as optical nonlinearity, (semi)conductivity, ferroelectricity, and magnetism. Although the properties of these cocrystals have been investigated for decades, the principal challenge that researchers face currently is to devise an efficient approach which allows for the growth of high-quality crystalline materials, in anticipation of a host of different technological applications. The research reported here introduces an innovative design, termed LASO?lock-arm supramolecular ordering?in the form of a modular approach for the development of responsive organic cocrystals. The strategy relies on the use of aromatic electronic donor and acceptor building blocks, carrying complementary rigid and flexible arms, capable of forming hydrogen bonds to amplify the cocrystallization processes. The cooperativity of charge transfer and hydrogen-bonding interactions between the building blocks leads to binary cocrystals that have alternating donors and acceptors extending in one and two dimensions sustained by an intricate network of hydrogen bonds. A variety of air-stable, mechanically robust, centimeter-long, organic charge transfer cocrystals have been grown by liquid?liquid diffusion under ambient conditions inside 72 h. These cocrystals are of considerable interest because of their remarkable size and stability and the promise they hold when it comes to fabricating the next generation of innovative electronic and photonic devices.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Organic charge transfer cocrystals are inexpensive, modular, and solution-processable materials that are able, in some instances, to exhibit properties such as optical nonlinearity, (semi)conductivity, ferroelectricity, and magnetism. Although the properties of these cocrystals have been investigated for decades, the principal challenge that researchers face currently is to devise an efficient approach which allows for the growth of high-quality crystalline materials, in anticipation of a host of different technological applications. The research reported here introduces an innovative design, termed LASO?lock-arm supramolecular ordering?in the form of a modular approach for the development of responsive organic cocrystals. The strategy relies on the use of aromatic electronic donor and acceptor building blocks, carrying complementary rigid and flexible arms, capable of forming hydrogen bonds to amplify the cocrystallization processes. The cooperativity of charge transfer and hydrogen-bonding interactions between the building blocks leads to binary cocrystals that have alternating donors and acceptors extending in one and two dimensions sustained by an intricate network of hydrogen bonds. A variety of air-stable, mechanically robust, centimeter-long, organic charge transfer cocrystals have been grown by liquid?liquid diffusion under ambient conditions inside 72 h. These cocrystals are of considerable interest because of their remarkable size and stability and the promise they hold when it comes to fabricating the next generation of innovative electronic and photonic devices.
Frasconi, Marco; Kikuchi, Takashi; Cao, Dennis; Wu, Yilei; Liu, Wei-Guang; Dyar, Scott M.; Barin, Gokhan; Sarjeant, Amy A.; Stern, Charlotte L.; Carmieli, Raanan; Wang, Cheng; Wasielewski, Michael R.; Goddard, William A.; Stoddart, J. Fraser
Mechanical Bonds and Topological Effects in Radical Dimer Stabilization Journal Article
In: J. Am. Chem. Soc., no. 136, pp. 11011−11026, 2014, ISSN: 0002-7863.
@article{frasconi_mechanical_2014,
title = {Mechanical Bonds and Topological Effects in Radical Dimer Stabilization},
author = {Frasconi, Marco and Kikuchi, Takashi and Cao, Dennis and Wu, Yilei and Liu, Wei-Guang and Dyar, Scott M. and Barin, Gokhan and Sarjeant, Amy A. and Stern, Charlotte L. and Carmieli, Raanan and Wang, Cheng and Wasielewski, Michael R. and Goddard, William A. and Stoddart, J. Fraser},
url = {http://dx.doi.org/10.1021/ja504662a},
doi = {10.1021/ja504662a},
issn = {0002-7863},
year = {2014},
date = {2014-07-01},
urldate = {2014-08-04},
journal = {J. Am. Chem. Soc.},
number = {136},
pages = {11011−11026},
abstract = {While mechanical bonding stabilizes tetrathiafulvalene (TTF) radical dimers, the question arises: what role does topology play in catenanes containing TTF units? Here, we report how topology, together with mechanical bonding, in isomeric [3]- and doubly interlocked [2]catenanes controls the formation of TTF radical dimers within their structural frameworks, including a ring-in-ring complex (formed between an organoplatinum square and a 2+2 macrocyclic polyether containing two 1,5-dioxynaphthalene (DNP) and two TTF units) that is topologically isomeric with the doubly interlocked [2]catenane. The separate TTF units in the two 1+1 macrocycles (each containing also one DNP unit) of the isomeric [3]catenane exhibit slightly different redox properties compared with those in the 2+2 macrocycle present in the [2]catenane, while comparison with its topological isomer reveals substantially different redox behavior. Although the stabilities of the mixed-valence (TTF2)?+ dimers are similar in the two catenanes, the radical cationic (TTF?+)2 dimer in the [2]catenane occurs only fleetingly compared with its prominent existence in the [3]catenane, while both dimers are absent altogether in the ring-in-ring complex. The electrochemical behavior of these three radically configurable isomers demonstrates that a fundamental relationship exists between topology and redox properties.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
While mechanical bonding stabilizes tetrathiafulvalene (TTF) radical dimers, the question arises: what role does topology play in catenanes containing TTF units? Here, we report how topology, together with mechanical bonding, in isomeric [3]- and doubly interlocked [2]catenanes controls the formation of TTF radical dimers within their structural frameworks, including a ring-in-ring complex (formed between an organoplatinum square and a 2+2 macrocyclic polyether containing two 1,5-dioxynaphthalene (DNP) and two TTF units) that is topologically isomeric with the doubly interlocked [2]catenane. The separate TTF units in the two 1+1 macrocycles (each containing also one DNP unit) of the isomeric [3]catenane exhibit slightly different redox properties compared with those in the 2+2 macrocycle present in the [2]catenane, while comparison with its topological isomer reveals substantially different redox behavior. Although the stabilities of the mixed-valence (TTF2)?+ dimers are similar in the two catenanes, the radical cationic (TTF?+)2 dimer in the [2]catenane occurs only fleetingly compared with its prominent existence in the [3]catenane, while both dimers are absent altogether in the ring-in-ring complex. The electrochemical behavior of these three radically configurable isomers demonstrates that a fundamental relationship exists between topology and redox properties.
Cao, Dennis; Hong, Michael; Blackburn, Anthea K.; Liu, Zhichang; Holcroft, James M.; Stoddart, J. Fraser
Two-point Halogen Bonding between 3,6-Dihalopyromellitic Diimides Journal Article
In: Chem. Sci., no. 5, pp. 4242−4248, 2014, ISSN: 2041-6539.
@article{cao_two-point_2014,
title = {Two-point Halogen Bonding between 3,6-Dihalopyromellitic Diimides},
author = {Cao, Dennis and Hong, Michael and Blackburn, Anthea K. and Liu, Zhichang and Holcroft, James M. and Stoddart, J. Fraser},
url = {http://pubs.rsc.org/en/content/articlelanding/2014/sc/c4sc00999a},
doi = {10.1039/C4SC00999A},
issn = {2041-6539},
year = {2014},
date = {2014-07-01},
urldate = {2014-08-04},
journal = {Chem. Sci.},
number = {5},
pages = {4242−4248},
abstract = {The syntheses of 3,6-dichloro-, -dibromo-, and -diiodopyromellitic diimides—ACl, ABr, and AI, respectively—have been achieved. X-Ray crystallography of single crystals of ACl and ABr unveils the formation of extensive halogen-bonding networks in the solid-state as a consequence of interactions between the lone pairs on the carbonyl oxygen atoms with the σ-holes of the halogen atoms. Further, the solid-state superstructure of diiodopyromellitic diimide is characterised by the formation of associated halogen-π dimers. The co-crystallisation of ACl or ABr with a 1,5-diaminonaphthalene derivative DN yields co-crystals of a mixed-stack charge-transfer (CT) complex which are supported by an expansive hydrogen-bonded network in addition to halogen-bonded belts that bring adjacent mixed-stacks into association with each other. 2,6-Dimethoxynaphthalene (DO) proved to be an effective CT complement to AI, yielding solvent-free co-crystals with superstructures which are comprised of a 1:2 ratio of AI to DO. This dimeric halogen-bonding motif is reminiscent of the formation of hydrogen-bonded dimers between carboxylic acids.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The syntheses of 3,6-dichloro-, -dibromo-, and -diiodopyromellitic diimides—ACl, ABr, and AI, respectively—have been achieved. X-Ray crystallography of single crystals of ACl and ABr unveils the formation of extensive halogen-bonding networks in the solid-state as a consequence of interactions between the lone pairs on the carbonyl oxygen atoms with the σ-holes of the halogen atoms. Further, the solid-state superstructure of diiodopyromellitic diimide is characterised by the formation of associated halogen-π dimers. The co-crystallisation of ACl or ABr with a 1,5-diaminonaphthalene derivative DN yields co-crystals of a mixed-stack charge-transfer (CT) complex which are supported by an expansive hydrogen-bonded network in addition to halogen-bonded belts that bring adjacent mixed-stacks into association with each other. 2,6-Dimethoxynaphthalene (DO) proved to be an effective CT complement to AI, yielding solvent-free co-crystals with superstructures which are comprised of a 1:2 ratio of AI to DO. This dimeric halogen-bonding motif is reminiscent of the formation of hydrogen-bonded dimers between carboxylic acids.
Liu, Zhichang; Lei, Juying; Frasconi, Marco; Li, Xiaohu; Cao, Dennis; Zhu, Zhixue; Schneebeli, Severin T.; Schatz, George C.; Stoddart, J. Fraser
A Square-Planar Tetracoordinate Oxygen-Containing Ti4O17 Cluster Stabilized by Two 1,1′-Ferrocenedicarboxylato Ligands Journal Article
In: Angew. Chem. Int. Ed., pp. 9193−9197, 2014, ISSN: 1521-3773.
@article{liu_square-planar_2014,
title = {A Square-Planar Tetracoordinate Oxygen-Containing Ti4O17 Cluster Stabilized by Two 1,1′-Ferrocenedicarboxylato Ligands},
author = {Liu, Zhichang and Lei, Juying and Frasconi, Marco and Li, Xiaohu and Cao, Dennis and Zhu, Zhixue and Schneebeli, Severin T. and Schatz, George C. and Stoddart, J. Fraser},
url = {http://onlinelibrary.wiley.com/doi/10.1002/anie.201402603/abstract},
doi = {10.1002/anie.201402603},
issn = {1521-3773},
year = {2014},
date = {2014-01-01},
urldate = {2014-08-04},
journal = {Angew. Chem. Int. Ed.},
pages = {9193−9197},
abstract = {By introducing steric constraints into molecular compounds, it is possible to achieve atypical coordination geometries for the elements. Herein, we demonstrate that a titanium-oxo cluster [Ti4(μ4-O)(μ2-O)2(OPri)6(fdc)2], which possesses a unique edge-sharing Ti4O17 octahedron tetramer core, is stabilized by the constraints produced by two orthogonal 1,1′-ferrocenedicarboxylato (fdc) ligands. As a result, a square-planar tetracoordinate oxygen (ptO) can be generated. The bonding pattern of this unusual anti-van’t Hoff/Le Bel oxygen, which has been probed by theoretical calculations, can be described by two horizontally σ-bonded 2px and 2py orbitals along with one perpendicular nonbonded 2pz orbital. While the two ferrocene units are separated spatially by the ptO with an Fe⋅⋅⋅Fe separation of 10.4 Å, electronic communication between them still takes place as revealed by the cluster’s two distinct one-electron electrochemical oxidation processes.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
By introducing steric constraints into molecular compounds, it is possible to achieve atypical coordination geometries for the elements. Herein, we demonstrate that a titanium-oxo cluster [Ti4(μ4-O)(μ2-O)2(OPri)6(fdc)2], which possesses a unique edge-sharing Ti4O17 octahedron tetramer core, is stabilized by the constraints produced by two orthogonal 1,1′-ferrocenedicarboxylato (fdc) ligands. As a result, a square-planar tetracoordinate oxygen (ptO) can be generated. The bonding pattern of this unusual anti-van’t Hoff/Le Bel oxygen, which has been probed by theoretical calculations, can be described by two horizontally σ-bonded 2px and 2py orbitals along with one perpendicular nonbonded 2pz orbital. While the two ferrocene units are separated spatially by the ptO with an Fe⋅⋅⋅Fe separation of 10.4 Å, electronic communication between them still takes place as revealed by the cluster’s two distinct one-electron electrochemical oxidation processes.
2013
Hartlieb, Karel J.; Blackburn, Anthea K.; Schneebeli, Severin T.; Forgan, Ross S.; Sarjeant, Amy A.; Stern, Charlotte L.; Cao, Dennis; Stoddart, J. Fraser
Topological Isomerism in a Chiral Handcuff Catenane Journal Article
In: Chem. Sci., vol. 5, no. 1, pp. 90−100, 2013, ISSN: 2041-6539.
@article{hartlieb_topological_2013,
title = {Topological Isomerism in a Chiral Handcuff Catenane},
author = {Hartlieb, Karel J. and Blackburn, Anthea K. and Schneebeli, Severin T. and Forgan, Ross S. and Sarjeant, Amy A. and Stern, Charlotte L. and Cao, Dennis and Stoddart, J. Fraser},
url = {http://pubs.rsc.org/en/content/articlelanding/2014/sc/c3sc52106k},
doi = {10.1039/C3SC52106K},
issn = {2041-6539},
year = {2013},
date = {2013-11-01},
urldate = {2014-04-01},
journal = {Chem. Sci.},
volume = {5},
number = {1},
pages = {90−100},
abstract = {The self-assembly of two topological isomers of a handcuff catenane has been achieved by utilizing the template-directed synthesis between the π-electron-rich bis-1,5-dioxynaphtho[50]crown-14 and the precursors to two fused π-electron-deficient cyclobis(paraquat-p-phenylene) cyclophanes. Characterization of the product using 1H NMR spectroscopy and single-crystal X-ray diffraction, with supporting density functional theory (DFT) calculations, suggests that the 1,5-dioxynaphthalene units in the major topological isomer align themselves with the same relative orientations inside the cyclophanes on account of restrictions imposed by the lengths of the polyether loops. The DFT calculations also reveal that the energies of the two topological isomers are similar to each other, supporting the experimental observation that both isomers can be isolated as a mixture from a one-pot reaction. The two isomers – designated as the meta–meta and ortho–ortho isomers with different topologies that are not interconvertible – only differ in the manner in which the polyether loops wind their way around the central 1,2,4,5-tetrasubstituted benzenoid ring in the ditopic host. X-Ray crystallography proves that by far the major topological isomer in the solid state is the meta–meta one. 1H NMR spectroscopy confirms that it is also the major isomer in solution, whilst also revealing the presence of a minor isomer, which is assumed, for the time-being, to have the ortho–ortho topology. The free fused ditopic host has been obtained using a protocol similar to that employed in the template-directed synthesis of the handcuff catenane, except that the crown ether is replaced with an acyclic template which can be removed post-synthesis. The results of isothermal titration calorimetry studies shed some light on the mechanism of binding of π-electron-rich guests: they lead us to believe that when two electron-rich guests bind to the ditopic host, they do so with allosteric negative cooperativity.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The self-assembly of two topological isomers of a handcuff catenane has been achieved by utilizing the template-directed synthesis between the π-electron-rich bis-1,5-dioxynaphtho[50]crown-14 and the precursors to two fused π-electron-deficient cyclobis(paraquat-p-phenylene) cyclophanes. Characterization of the product using 1H NMR spectroscopy and single-crystal X-ray diffraction, with supporting density functional theory (DFT) calculations, suggests that the 1,5-dioxynaphthalene units in the major topological isomer align themselves with the same relative orientations inside the cyclophanes on account of restrictions imposed by the lengths of the polyether loops. The DFT calculations also reveal that the energies of the two topological isomers are similar to each other, supporting the experimental observation that both isomers can be isolated as a mixture from a one-pot reaction. The two isomers – designated as the meta–meta and ortho–ortho isomers with different topologies that are not interconvertible – only differ in the manner in which the polyether loops wind their way around the central 1,2,4,5-tetrasubstituted benzenoid ring in the ditopic host. X-Ray crystallography proves that by far the major topological isomer in the solid state is the meta–meta one. 1H NMR spectroscopy confirms that it is also the major isomer in solution, whilst also revealing the presence of a minor isomer, which is assumed, for the time-being, to have the ortho–ortho topology. The free fused ditopic host has been obtained using a protocol similar to that employed in the template-directed synthesis of the handcuff catenane, except that the crown ether is replaced with an acyclic template which can be removed post-synthesis. The results of isothermal titration calorimetry studies shed some light on the mechanism of binding of π-electron-rich guests: they lead us to believe that when two electron-rich guests bind to the ditopic host, they do so with allosteric negative cooperativity.
Cao, Dennis; Juríček, Michal; Brown, Zachary J.; Sue, Andrew C. -H.; Liu, Zhichang; Lei, Juying; Blackburn, Anthea K.; Grunder, Sergio; Sarjeant, Amy A.; Coskun, Ali; Wang, Cheng; Farha, Omar K.; Hupp, Joseph T.; Stoddart, J. Fraser
Three-Dimensional Architectures Incorporating Stereoregular Donor–Acceptor Stacks Journal Article
In: Chem. Eur. J., vol. 19, no. 26, pp. 8457−8465, 2013, ISSN: 1521-3765.
@article{cao_three-dimensional_2013,
title = {Three-Dimensional Architectures Incorporating Stereoregular Donor–Acceptor Stacks},
author = {Cao, Dennis and Juríček, Michal and Brown, Zachary J. and Sue, Andrew C.-H. and Liu, Zhichang and Lei, Juying and Blackburn, Anthea K. and Grunder, Sergio and Sarjeant, Amy A. and Coskun, Ali and Wang, Cheng and Farha, Omar K. and Hupp, Joseph T. and Stoddart, J. Fraser},
url = {http://onlinelibrary.wiley.com/doi/10.1002/chem.201300762/abstract},
doi = {10.1002/chem.201300762},
issn = {1521-3765},
year = {2013},
date = {2013-06-01},
urldate = {2014-03-25},
journal = {Chem. Eur. J.},
volume = {19},
number = {26},
pages = {8457−8465},
abstract = {We report the synthesis of two [2]catenane-containing struts that are composed of a tetracationic cyclophane (TC4+) encircling a 1,5-dioxynaphthalene (DNP)-based crown ether, which bears two terphenylene arms. The TC4+ rings comprise either 1) two bipyridinium (BIPY2+) units or 2) a BIPY2+ and a diazapyrenium (DAP2+) unit. These degenerate and nondegenerate catenanes were reacted in the presence of Cu(NO3)2⋅2.5 H2O to yield Cu-paddlewheel-based MOF-1050 and MOF-1051. The solid-state structures of these MOFs reveal that the metal clusters serve to join the heptaphenylene struts into grid-like 2D networks. These 2D sheets are then held together by infinite donor–acceptor stacks involving the [2]catenanes to produce interpenetrated 3D architectures. As a consequence of the planar chirality associated with both the DNP and hydroquinone (HQ) units present in the crown ether, each catenane can exist as four stereoisomers. In the case of the nondegenerate (bistable) catenane, the situation is further complicated by the presence of translational isomers. Upon crystallization, however, only two of the four possible stereoisomers—namely, the enantiomeric RR and SS forms—are observed in the crystals. An additional element of co-conformational selectivity is present in MOF-1051 as a consequence of the substitution of one of the BIPY2+ units by a DAP2+ unit: only the translational isomer in which the DAP2+ unit is encircled by the crown ether is observed. The overall topologies of MOF-1050 and MOF-1051, and the selective formation of stereoisomers and translational isomers during the kinetically driven crystallization, provide evidence that weak noncovalent bonding interactions play a significant role in the assembly of these extended (super)structures.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
We report the synthesis of two [2]catenane-containing struts that are composed of a tetracationic cyclophane (TC4+) encircling a 1,5-dioxynaphthalene (DNP)-based crown ether, which bears two terphenylene arms. The TC4+ rings comprise either 1) two bipyridinium (BIPY2+) units or 2) a BIPY2+ and a diazapyrenium (DAP2+) unit. These degenerate and nondegenerate catenanes were reacted in the presence of Cu(NO3)2⋅2.5 H2O to yield Cu-paddlewheel-based MOF-1050 and MOF-1051. The solid-state structures of these MOFs reveal that the metal clusters serve to join the heptaphenylene struts into grid-like 2D networks. These 2D sheets are then held together by infinite donor–acceptor stacks involving the [2]catenanes to produce interpenetrated 3D architectures. As a consequence of the planar chirality associated with both the DNP and hydroquinone (HQ) units present in the crown ether, each catenane can exist as four stereoisomers. In the case of the nondegenerate (bistable) catenane, the situation is further complicated by the presence of translational isomers. Upon crystallization, however, only two of the four possible stereoisomers—namely, the enantiomeric RR and SS forms—are observed in the crystals. An additional element of co-conformational selectivity is present in MOF-1051 as a consequence of the substitution of one of the BIPY2+ units by a DAP2+ unit: only the translational isomer in which the DAP2+ unit is encircled by the crown ether is observed. The overall topologies of MOF-1050 and MOF-1051, and the selective formation of stereoisomers and translational isomers during the kinetically driven crystallization, provide evidence that weak noncovalent bonding interactions play a significant role in the assembly of these extended (super)structures.
Liu, Zhichang; Frasconi, Marco; Lei, Juying; Brown, Zachary J.; Zhu, Zhixue; Cao, Dennis; Iehl, Julien; Liu, Guoliang; Fahrenbach, Albert C.; Botros, Youssry Y.; Farha, Omar K.; Hupp, Joseph T.; Mirkin, Chad A.; Fraser Stoddart, J.
Selective Isolation of Gold Facilitated by Second-Sphere Coordination with α-Cyclodextrin Journal Article
In: Nature Commun., vol. 4, pp. 1855, 2013, ISSN: 2041-1733.
@article{liu_selective_2013,
title = {Selective Isolation of Gold Facilitated by Second-Sphere Coordination with α-Cyclodextrin},
author = {Liu, Zhichang and Frasconi, Marco and Lei, Juying and Brown, Zachary J. and Zhu, Zhixue and Cao, Dennis and Iehl, Julien and Liu, Guoliang and Fahrenbach, Albert C. and Botros, Youssry Y. and Farha, Omar K. and Hupp, Joseph T. and Mirkin, Chad A. and Fraser Stoddart, J.},
url = {http://www.nature.com/ncomms/journal/v4/n5/full/ncomms2891.html},
doi = {10.1038/ncomms2891},
issn = {2041-1733},
year = {2013},
date = {2013-01-01},
urldate = {2013-05-17},
journal = {Nature Commun.},
volume = {4},
pages = {1855},
abstract = {Gold recovery using environmentally benign chemistry is imperative from an environmental perspective. Here we report the spontaneous assembly of a one-dimensional supramolecular complex with an extended [K(OH2)6][AuBr4] (α-cyclodextrin)2n chain superstructure formed during the rapid co-precipitation of α-cyclodextrin and KAuBr4 in water. This phase change is selective for this gold salt, even in the presence of other square-planar palladium and platinum complexes. From single-crystal X-ray analyses of six inclusion complexes between α-, β- and γ-cyclodextrins with KAuBr4 and KAuCl4, we hypothesize that a perfect match in molecular recognition between α-cyclodextrin and [AuBr4]− leads to a near-axial orientation of the ion with respect to the α-cyclodextrin channel, which facilitates a highly specific second-sphere coordination involving [AuBr4]− and [K(OH2)6]+ and drives the co-precipitation of the 1:2 adduct. This discovery heralds a green host–guest procedure for gold recovery from gold-bearing raw materials making use of α-cyclodextrin—an inexpensive and environmentally benign carbohydrate.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Gold recovery using environmentally benign chemistry is imperative from an environmental perspective. Here we report the spontaneous assembly of a one-dimensional supramolecular complex with an extended [K(OH2)6][AuBr4] (α-cyclodextrin)2n chain superstructure formed during the rapid co-precipitation of α-cyclodextrin and KAuBr4 in water. This phase change is selective for this gold salt, even in the presence of other square-planar palladium and platinum complexes. From single-crystal X-ray analyses of six inclusion complexes between α-, β- and γ-cyclodextrins with KAuBr4 and KAuCl4, we hypothesize that a perfect match in molecular recognition between α-cyclodextrin and [AuBr4]− leads to a near-axial orientation of the ion with respect to the α-cyclodextrin channel, which facilitates a highly specific second-sphere coordination involving [AuBr4]− and [K(OH2)6]+ and drives the co-precipitation of the 1:2 adduct. This discovery heralds a green host–guest procedure for gold recovery from gold-bearing raw materials making use of α-cyclodextrin—an inexpensive and environmentally benign carbohydrate.
Barnes, Jonathan C.; Fahrenbach, Albert C.; Cao, Dennis; Dyar, Scott M.; Frasconi, Marco; Giesener, Marc A.; Benítez, Diego; Tkatchouk, Ekaterina; Chernyashevskyy, Oleksandr; Shin, Weon Ho; Li, Hao; Sampath, Srinivasan; Stern, Charlotte L.; Sarjeant, Amy A.; Hartlieb, Karel J.; Liu, Zhichang; Carmieli, Raanan; Botros, Youssry Y.; Choi, Jang Wook; Slawin, Alexandra M. Z.; Ketterson, John B.; Wasielewski, Michael R.; Goddard, William A.; Stoddart, J. Fraser
A Radically Configurable Six-State Compound Journal Article
In: Science, vol. 339, no. 6118, pp. 429−433, 2013, ISSN: 0036-8075, 1095-9203.
@article{barnes_radically_2013,
title = {A Radically Configurable Six-State Compound},
author = {Barnes, Jonathan C. and Fahrenbach, Albert C. and Cao, Dennis and Dyar, Scott M. and Frasconi, Marco and Giesener, Marc A. and Benítez, Diego and Tkatchouk, Ekaterina and Chernyashevskyy, Oleksandr and Shin, Weon Ho and Li, Hao and Sampath, Srinivasan and Stern, Charlotte L. and Sarjeant, Amy A. and Hartlieb, Karel J. and Liu, Zhichang and Carmieli, Raanan and Botros, Youssry Y. and Choi, Jang Wook and Slawin, Alexandra M. Z. and Ketterson, John B. and Wasielewski, Michael R. and Goddard, William A. and Stoddart, J. Fraser},
url = {http://www.sciencemag.org/content/339/6118/429},
doi = {10.1126/science.1228429},
issn = {0036-8075, 1095-9203},
year = {2013},
date = {2013-01-01},
urldate = {2013-05-17},
journal = {Science},
volume = {339},
number = {6118},
pages = {429−433},
abstract = {Most organic radicals possess short lifetimes and quickly undergo dimerization or oxidation. Here, we report on the synthesis by radical templation of a class of air- and water-stable organic radicals, trapped within a homo[2]catenane composed of two rigid and fixed cyclobis(paraquat-p-phenylene) rings. The highly energetic octacationic homo[2]catenane, which is capable of accepting up to eight electrons, can be configured reversibly, both chemically and electrochemically, between each one of six experimentally accessible redox states (0, 2+, 4+, 6+, 7+, and 8+) from within the total of nine states evaluated by quantum mechanical methods. All six of the observable redox states have been identified by electrochemical techniques, three (4+, 6+, and 7+) have been characterized by x-ray crystallography, four (4+, 6+, 7+, and 8+) by electron paramagnetic resonance spectroscopy, one (7+) by superconducting quantum interference device magnetometry, and one (8+) by nuclear magnetic resonance spectroscopy.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Most organic radicals possess short lifetimes and quickly undergo dimerization or oxidation. Here, we report on the synthesis by radical templation of a class of air- and water-stable organic radicals, trapped within a homo[2]catenane composed of two rigid and fixed cyclobis(paraquat-p-phenylene) rings. The highly energetic octacationic homo[2]catenane, which is capable of accepting up to eight electrons, can be configured reversibly, both chemically and electrochemically, between each one of six experimentally accessible redox states (0, 2+, 4+, 6+, 7+, and 8+) from within the total of nine states evaluated by quantum mechanical methods. All six of the observable redox states have been identified by electrochemical techniques, three (4+, 6+, and 7+) have been characterized by x-ray crystallography, four (4+, 6+, 7+, and 8+) by electron paramagnetic resonance spectroscopy, one (7+) by superconducting quantum interference device magnetometry, and one (8+) by nuclear magnetic resonance spectroscopy.
Jia, Chuancheng; Li, Hao; Jiang, Jiaolong; Wang, Jindong; Chen, Hongliang; Cao, Dennis; Stoddart, J. Fraser; Guo, Xuefeng
Interface-Engineered Bistable [2]Rotaxane-Graphene Hybrids with Logic Capabilities Journal Article
In: Adv. Mater., vol. 25, no. 46, pp. 6752−6759, 2013, ISSN: 1521-4095.
@article{jia_interface-engineered_2013,
title = {Interface-Engineered Bistable [2]Rotaxane-Graphene Hybrids with Logic Capabilities},
author = {Jia, Chuancheng and Li, Hao and Jiang, Jiaolong and Wang, Jindong and Chen, Hongliang and Cao, Dennis and Stoddart, J. Fraser and Guo, Xuefeng},
url = {http://onlinelibrary.wiley.com.turing.library.northwestern.edu/doi/10.1002/adma.201302393/abstract},
doi = {10.1002/adma.201302393},
issn = {1521-4095},
year = {2013},
date = {2013-01-01},
urldate = {2014-05-28},
journal = {Adv. Mater.},
volume = {25},
number = {46},
pages = {6752−6759},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2012
Xue, Min; Cao, Dennis; Stoddart, J. Fraser; Zink, Jeffrey I.
Size-Selective pH-Operated Megagates on Mesoporous Silica Materials Journal Article
In: Nanoscale, vol. 4, no. 23, pp. 7569−7574, 2012, ISSN: 2040-3372.
@article{xue_size-selective_2012,
title = {Size-Selective pH-Operated Megagates on Mesoporous Silica Materials},
author = {Xue, Min and Cao, Dennis and Stoddart, J. Fraser and Zink, Jeffrey I.},
url = {http://pubs.rsc.org/en/content/articlelanding/2012/nr/c2nr32170j},
doi = {10.1039/C2NR32170J},
issn = {2040-3372},
year = {2012},
date = {2012-11-01},
urldate = {2013-05-17},
journal = {Nanoscale},
volume = {4},
number = {23},
pages = {7569−7574},
abstract = {pH-responsive megagates have been fabricated around mesoporous silica material SBA-15 in order to mechanize the mesopores. These megagates remain closed in neutral conditions, but open at pH 5. The capping components of the megagates were designed to be capable of controlling pores up to 6.5 nm in diameter.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
pH-responsive megagates have been fabricated around mesoporous silica material SBA-15 in order to mechanize the mesopores. These megagates remain closed in neutral conditions, but open at pH 5. The capping components of the megagates were designed to be capable of controlling pores up to 6.5 nm in diameter.
Tayi, Alok S.; Shveyd, Alexander K.; Sue, Andrew C. -H.; Szarko, Jodi M.; Rolczynski, Brian S.; Cao, Dennis; Kennedy, T. Jackson; Sarjeant, Amy A.; Stern, Charlotte L.; Paxton, Walter F.; Wu, Wei; Dey, Sanjeev K.; Fahrenbach, Albert C.; Guest, Jeffrey R.; Mohseni, Hooman; Chen, Lin X.; Wang, Kang L.; Stoddart, J. Fraser; Stupp, Samuel I.
Room-Temperature Ferroelectricity in Supramolecular Networks of Charge-Transfer Complexes Journal Article
In: Nature, vol. 488, no. 7412, pp. 485−489, 2012, ISSN: 0028-0836.
@article{tayi_room-temperature_2012,
title = {Room-Temperature Ferroelectricity in Supramolecular Networks of Charge-Transfer Complexes},
author = {Tayi, Alok S. and Shveyd, Alexander K. and Sue, Andrew C.-H. and Szarko, Jodi M. and Rolczynski, Brian S. and Cao, Dennis and Kennedy, T. Jackson and Sarjeant, Amy A. and Stern, Charlotte L. and Paxton, Walter F. and Wu, Wei and Dey, Sanjeev K. and Fahrenbach, Albert C. and Guest, Jeffrey R. and Mohseni, Hooman and Chen, Lin X. and Wang, Kang L. and Stoddart, J. Fraser and Stupp, Samuel I.},
url = {http://www.nature.com/nature/journal/v488/n7412/full/nature11395.html},
doi = {10.1038/nature11395},
issn = {0028-0836},
year = {2012},
date = {2012-08-01},
urldate = {2013-05-17},
journal = {Nature},
volume = {488},
number = {7412},
pages = {485−489},
abstract = {Materials exhibiting a spontaneous electrical polarization that can be switched easily between antiparallel orientations are of potential value for sensors, photonics and energy-efficient memories. In this context, organic ferroelectrics are of particular interest because they promise to be lightweight, inexpensive and easily processed into devices. A recently identified family of organic ferroelectric structures is based on intermolecular charge transfer, where donor and acceptor molecules co-crystallize in an alternating fashion known as a mixed stack: in the crystalline lattice, a collective transfer of electrons from donor to acceptor molecules results in the formation of dipoles that can be realigned by an external field as molecules switch partners in the mixed stack. Although mixed stacks have been investigated extensively, only three systems are known to show ferroelectric switching, all below 71 kelvin. Here we describe supramolecular charge-transfer networks that undergo ferroelectric polarization switching with a ferroelectric Curie temperature above room temperature. These polar and switchable systems utilize a structural synergy between a hydrogen-bonded network and charge-transfer complexation of donor and acceptor molecules in a mixed stack. This supramolecular motif could help guide the development of other functional organic systems that can switch polarization under the influence of electric fields at ambient temperatures.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Materials exhibiting a spontaneous electrical polarization that can be switched easily between antiparallel orientations are of potential value for sensors, photonics and energy-efficient memories. In this context, organic ferroelectrics are of particular interest because they promise to be lightweight, inexpensive and easily processed into devices. A recently identified family of organic ferroelectric structures is based on intermolecular charge transfer, where donor and acceptor molecules co-crystallize in an alternating fashion known as a mixed stack: in the crystalline lattice, a collective transfer of electrons from donor to acceptor molecules results in the formation of dipoles that can be realigned by an external field as molecules switch partners in the mixed stack. Although mixed stacks have been investigated extensively, only three systems are known to show ferroelectric switching, all below 71 kelvin. Here we describe supramolecular charge-transfer networks that undergo ferroelectric polarization switching with a ferroelectric Curie temperature above room temperature. These polar and switchable systems utilize a structural synergy between a hydrogen-bonded network and charge-transfer complexation of donor and acceptor molecules in a mixed stack. This supramolecular motif could help guide the development of other functional organic systems that can switch polarization under the influence of electric fields at ambient temperatures.
Wang, Cheng; Dyar, Scott M.; Cao, Dennis; Fahrenbach, Albert C.; Horwitz, Noah; Colvin, Michael T.; Carmieli, Raanan; Stern, Charlotte L.; Dey, Sanjeev K.; Wasielewski, Michael R.; Stoddart, J. Fraser
Tetrathiafulvalene Hetero Radical Cation Dimerization in a Redox-Active [2]Catenane Journal Article
In: J. Am. Chem. Soc., vol. 134, no. 46, pp. 19136−19145, 2012, ISSN: 0002-7863.
@article{wang_tetrathiafulvalene_2012,
title = {Tetrathiafulvalene Hetero Radical Cation Dimerization in a Redox-Active [2]Catenane},
author = {Wang, Cheng and Dyar, Scott M. and Cao, Dennis and Fahrenbach, Albert C. and Horwitz, Noah and Colvin, Michael T. and Carmieli, Raanan and Stern, Charlotte L. and Dey, Sanjeev K. and Wasielewski, Michael R. and Stoddart, J. Fraser},
url = {http://dx.doi.org/10.1021/ja307577t},
doi = {10.1021/ja307577t},
issn = {0002-7863},
year = {2012},
date = {2012-01-01},
urldate = {2013-05-17},
journal = {J. Am. Chem. Soc.},
volume = {134},
number = {46},
pages = {19136−19145},
abstract = {The electronic properties of tetrathiafulvalene (TTF) can be tuned by attaching electron-donating or electron-withdrawing substituents. An electron-rich macrocyclic polyether containing two TTF units of different constitutions, namely 4,4?-bis(hydroxymethyl)tetrathiafulvalene (OTTFO) and 4,4?-bisthiotetrathiafulvalene (STTFS), has been synthesized. On two-electron oxidation, a hetero radical dimer is formed between OTTFO?+ and STTFS?+. The redox behavior of the macrocyclic polyether has been investigated by electrochemical techniques and UV?vis and electron paramagnetic resonance (EPR) spectroscopies. The [2]catenane in which the macrocyclic polyether is mechanically interlocked with the cyclobis(paraquat-p-phenylene) (CBPQT4+) ring has also been prepared using template-directed protocols. In the case of the [2]catenane, the formation of the TTF hetero radical dimer is prevented sterically by the CBPQT4+ ring. After a one-electron oxidation, a 70:30 ratio of OTTFO?+ to STTFS?+ is present at equilibrium, and, as a result, two translational isomers of the [2]catenane associated with these electronically different isomeric states transpire. EPR titration spectroscopy and simulations reveal that the radical states of the two constitutionally different TTF units in the [2]catenane still experience long-range electronic intramolecular coupling interactions, despite the presence of the CBPQT4+ ring, when one or both of them are oxidized to the radical cationic state. These findings in the case of both the free macrocyclic polyether and the [2]catenane have led to a deeper fundamental understanding of the mechanism of radical cation dimer formation between constitutionally different TTF units.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The electronic properties of tetrathiafulvalene (TTF) can be tuned by attaching electron-donating or electron-withdrawing substituents. An electron-rich macrocyclic polyether containing two TTF units of different constitutions, namely 4,4?-bis(hydroxymethyl)tetrathiafulvalene (OTTFO) and 4,4?-bisthiotetrathiafulvalene (STTFS), has been synthesized. On two-electron oxidation, a hetero radical dimer is formed between OTTFO?+ and STTFS?+. The redox behavior of the macrocyclic polyether has been investigated by electrochemical techniques and UV?vis and electron paramagnetic resonance (EPR) spectroscopies. The [2]catenane in which the macrocyclic polyether is mechanically interlocked with the cyclobis(paraquat-p-phenylene) (CBPQT4+) ring has also been prepared using template-directed protocols. In the case of the [2]catenane, the formation of the TTF hetero radical dimer is prevented sterically by the CBPQT4+ ring. After a one-electron oxidation, a 70:30 ratio of OTTFO?+ to STTFS?+ is present at equilibrium, and, as a result, two translational isomers of the [2]catenane associated with these electronically different isomeric states transpire. EPR titration spectroscopy and simulations reveal that the radical states of the two constitutionally different TTF units in the [2]catenane still experience long-range electronic intramolecular coupling interactions, despite the presence of the CBPQT4+ ring, when one or both of them are oxidized to the radical cationic state. These findings in the case of both the free macrocyclic polyether and the [2]catenane have led to a deeper fundamental understanding of the mechanism of radical cation dimer formation between constitutionally different TTF units.
Cao, Dennis; Wang, Cheng; Giesener, Marc A.; Liu, Zhichang; Stoddart, J. Fraser
A Rigid Donor–Acceptor Daisy Chain Dimer Journal Article
In: Chem. Commun., vol. 48, no. 54, pp. 6791−6793, 2012, ISSN: 1364-548X.
@article{cao_rigid_2012,
title = {A Rigid Donor–Acceptor Daisy Chain Dimer},
author = {Cao, Dennis and Wang, Cheng and Giesener, Marc A. and Liu, Zhichang and Stoddart, J. Fraser},
url = {http://pubs.rsc.org/en/content/articlelanding/2012/cc/c2cc32499g},
doi = {10.1039/C2CC32499G},
issn = {1364-548X},
year = {2012},
date = {2012-01-01},
urldate = {2013-05-17},
journal = {Chem. Commun.},
volume = {48},
number = {54},
pages = {6791−6793},
abstract = {A functionalised cyclobis(paraquat-p-phenylene) attached by a rigid linker to a tetrathiafulvalene unit, which is incapable of self-complexation, forms preferentially a [c2]daisy chain which undergoes rapid disassociation and reassociation on the 1H NMR time-scale above room temperature. Aromaticity},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
A functionalised cyclobis(paraquat-p-phenylene) attached by a rigid linker to a tetrathiafulvalene unit, which is incapable of self-complexation, forms preferentially a [c2]daisy chain which undergoes rapid disassociation and reassociation on the 1H NMR time-scale above room temperature. Aromaticity
Fahrenbach, Albert C.; Bruns, Carson J.; Cao, Dennis; Stoddart, J. Fraser
Ground-State Thermodynamics of Bistable Redox-Active Donor–Acceptor Mechanically Interlocked Molecules Journal Article
In: Acc. Chem. Res., vol. 45, no. 9, pp. 1581−1592, 2012, ISSN: 0001-4842.
@article{fahrenbach_ground-state_2012,
title = {Ground-State Thermodynamics of Bistable Redox-Active Donor–Acceptor Mechanically Interlocked Molecules},
author = {Fahrenbach, Albert C. and Bruns, Carson J. and Cao, Dennis and Stoddart, J. Fraser},
url = {http://dx.doi.org/10.1021/ar3000629},
doi = {10.1021/ar3000629},
issn = {0001-4842},
year = {2012},
date = {2012-01-01},
urldate = {2013-05-17},
journal = {Acc. Chem. Res.},
volume = {45},
number = {9},
pages = {1581−1592},
abstract = {Fashioned through billions of years of evolution, biological molecular machines, such as ATP synthase, myosin, and kinesin, use the intricate relative motions of their components to drive some of life?s most essential processes. Having control over the motions in molecules is imperative for life to function, and many chemists have designed, synthesized, and investigated artificial molecular systems that also express controllable motions within molecules. Using bistable mechanically interlocked molecules (MIMs), based on donor?acceptor recognition motifs, we have sought to imitate the sophisticated nanoscale machines present in living systems. In this Account, we analyze the thermodynamic characteristics of a series of redox-switchable [2]rotaxanes and [2]catenanes. Control and understanding of the relative intramolecular movements of components in MIMs have been vital in the development of a variety of applications of these compounds ranging from molecular electronic devices to drug delivery systems. These bistable donor?acceptor MIMs undergo redox-activated switching between two isomeric states. Under ambient conditions, the dominant translational isomer, the ground-state coconformation (GSCC), is in equilibrium with the less favored translational isomer, the metastable-state coconformation (MSCC). By manipulating the redox state of the recognition site associated with the GSCC, we can stimulate the relative movements of the components in these bistable MIMs. The thermodynamic parameters of model host?guest complexes provide a good starting point to rationalize the ratio of GSCC to MSCC at equilibrium. The bistable [2]rotaxanes show a strong correlation between the relative free energies of model complexes and the ground-state distribution constants (KGS). This relationship does not always hold for bistable [2]catenanes, most likely because of the additional steric and electronic constraints present when the two rings are mechanically interlocked with each other. Measuring the ground-state distribution constants of bistable MIMs presents its own set of challenges. While it is possible, in principle, to determine these constants using NMR and UV?vis spectroscopies, these methods lack the sensitivity to permit the determination of ratios of translational isomers greater than 10:1 with sufficient accuracy and precision. A simple application of the Nernst equation, in combination with variable scan-rate cyclic voltammetry, however, allows the direct measurement of ground-state distribution constants across a wide range (KGS = 10?104) of values.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Fashioned through billions of years of evolution, biological molecular machines, such as ATP synthase, myosin, and kinesin, use the intricate relative motions of their components to drive some of life?s most essential processes. Having control over the motions in molecules is imperative for life to function, and many chemists have designed, synthesized, and investigated artificial molecular systems that also express controllable motions within molecules. Using bistable mechanically interlocked molecules (MIMs), based on donor?acceptor recognition motifs, we have sought to imitate the sophisticated nanoscale machines present in living systems. In this Account, we analyze the thermodynamic characteristics of a series of redox-switchable [2]rotaxanes and [2]catenanes. Control and understanding of the relative intramolecular movements of components in MIMs have been vital in the development of a variety of applications of these compounds ranging from molecular electronic devices to drug delivery systems. These bistable donor?acceptor MIMs undergo redox-activated switching between two isomeric states. Under ambient conditions, the dominant translational isomer, the ground-state coconformation (GSCC), is in equilibrium with the less favored translational isomer, the metastable-state coconformation (MSCC). By manipulating the redox state of the recognition site associated with the GSCC, we can stimulate the relative movements of the components in these bistable MIMs. The thermodynamic parameters of model host?guest complexes provide a good starting point to rationalize the ratio of GSCC to MSCC at equilibrium. The bistable [2]rotaxanes show a strong correlation between the relative free energies of model complexes and the ground-state distribution constants (KGS). This relationship does not always hold for bistable [2]catenanes, most likely because of the additional steric and electronic constraints present when the two rings are mechanically interlocked with each other. Measuring the ground-state distribution constants of bistable MIMs presents its own set of challenges. While it is possible, in principle, to determine these constants using NMR and UV?vis spectroscopies, these methods lack the sensitivity to permit the determination of ratios of translational isomers greater than 10:1 with sufficient accuracy and precision. A simple application of the Nernst equation, in combination with variable scan-rate cyclic voltammetry, however, allows the direct measurement of ground-state distribution constants across a wide range (KGS = 10?104) of values.
Wang, Cheng; Li, Zongxi; Cao, Dennis; Zhao, Yan-Li; Gaines, Justin W.; Bozdemir, O. Altan; Ambrogio, Michael W.; Frasconi, Marco; Botros, Youssry Y.; Zink, Jeffrey I.; Stoddart, J. Fraser
Stimulated Release of Size-Selected Cargos in Succession from Mesoporous Silica Nanoparticles Journal Article
In: Angew. Chem. Int. Ed., vol. 51, no. 22, pp. 5460−5465, 2012, ISSN: 1521-3773.
@article{wang_stimulated_2012,
title = {Stimulated Release of Size-Selected Cargos in Succession from Mesoporous Silica Nanoparticles},
author = {Wang, Cheng and Li, Zongxi and Cao, Dennis and Zhao, Yan-Li and Gaines, Justin W. and Bozdemir, O. Altan and Ambrogio, Michael W. and Frasconi, Marco and Botros, Youssry Y. and Zink, Jeffrey I. and Stoddart, J. Fraser},
url = {http://onlinelibrary.wiley.com/doi/10.1002/anie.201107960/abstract},
doi = {10.1002/anie.201107960},
issn = {1521-3773},
year = {2012},
date = {2012-01-01},
urldate = {2013-05-17},
journal = {Angew. Chem. Int. Ed.},
volume = {51},
number = {22},
pages = {5460−5465},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Wang, Cheng; Cao, Dennis; Fahrenbach, Albert C.; Grunder, Sergio; Dey, Sanjeev K.; Sarjeant, Amy A.; Stoddart, J. Fraser
The Effects of Conformation on the Noncovalent Bonding Interactions in a Bistable Donor–Acceptor [3]Catenane Journal Article
In: Chem. Commun., vol. 48, no. 74, pp. 9245−9247, 2012, ISSN: 1364-548X.
@article{wang_effects_2012,
title = {The Effects of Conformation on the Noncovalent Bonding Interactions in a Bistable Donor–Acceptor [3]Catenane},
author = {Wang, Cheng and Cao, Dennis and Fahrenbach, Albert C. and Grunder, Sergio and Dey, Sanjeev K. and Sarjeant, Amy A. and Stoddart, J. Fraser},
url = {http://pubs.rsc.org/en/content/articlelanding/2012/cc/c2cc34190e},
doi = {10.1039/C2CC34190E},
issn = {1364-548X},
year = {2012},
date = {2012-01-01},
urldate = {2013-05-17},
journal = {Chem. Commun.},
volume = {48},
number = {74},
pages = {9245−9247},
abstract = {A switchable donor–acceptor bistable [3]catenane, composed of a crown ether containing a pair of alternating π-electron rich tetrathiafulvalene and 1,5-dioxynaphthalene units, encircled by two π-electron deficient cyclobis(paraquat-p-phenylene) rings, has been synthesised and the redox-activated switching it},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
A switchable donor–acceptor bistable [3]catenane, composed of a crown ether containing a pair of alternating π-electron rich tetrathiafulvalene and 1,5-dioxynaphthalene units, encircled by two π-electron deficient cyclobis(paraquat-p-phenylene) rings, has been synthesised and the redox-activated switching it
Wang, Cheng; Cao, Dennis; Fahrenbach, Albert C.; Fang, Lei; Olson, Mark A.; Friedman, Douglas C.; Basu, Subhadeep; Dey, Sanjeev K.; Botros, Youssry Y.; Stoddart, J. Fraser
Solvent-Dependent Ground-State Distributions in a Donor–Acceptor Redox-Active Bistable [2]Catenane Journal Article
In: J. Phys. Org. Chem., vol. 25, no. 7, pp. 544−552, 2012, ISSN: 1099-1395.
@article{wang_solvent-dependent_2012,
title = {Solvent-Dependent Ground-State Distributions in a Donor–Acceptor Redox-Active Bistable [2]Catenane},
author = {Wang, Cheng and Cao, Dennis and Fahrenbach, Albert C. and Fang, Lei and Olson, Mark A. and Friedman, Douglas C. and Basu, Subhadeep and Dey, Sanjeev K. and Botros, Youssry Y. and Stoddart, J. Fraser},
url = {http://onlinelibrary.wiley.com/doi/10.1002/poc.1960/abstract?systemMessage=Wiley+Online+Library+will+be+disrupted+on+18+May+from+10%3A00-12%3A00+BST+%2805%3A00-07%3A00+EDT%29+for+essential+maintenance},
doi = {10.1002/poc.1960},
issn = {1099-1395},
year = {2012},
date = {2012-01-01},
urldate = {2013-05-17},
journal = {J. Phys. Org. Chem.},
volume = {25},
number = {7},
pages = {544−552},
abstract = {The solvent dependency of the ground-state distribution as well as the electrochemical switching behavior in a redox-active bistable donor–acceptor [2]catenane, containing bisthiotetrathiafulvalene (STTFS) and 1,5-dioxynaphthalene (DNP) recognition sites incorporated within a macrocyclic polyether encircled by the cyclobis(paraquat-p-phenylene) (CBPQT4+) ring, has been investigated. There are two translational isomers: (i) the ground-state co-conformation (GSCC) in which the CBPQT4+ ring encircles the STTFS unit and (ii) the metastable-state co-conformation (MSCC) in which the CBPQT4+ ring encircles the DNP unit. 1H NMR spectroscopy indicates that the ground-state distribution of GSCC to MSCC varies from approximately 1:1 in MeCN to 7:1 in MeCN : H2O (1:1, v/v) at 283 K. The reversible electrochemical switching behavior of the [2]catenane was confirmed by 1H NMR and UV−Vis spectroscopies, as well as by cyclic voltammetry (CV). Additionally, variable scan-rate CV studies were compared with simulated CV data and show that the ground-state distribution of GSCC to MSCC is about 30:1 in MeCN : H2O (1:1, v/v) at 298 K. With the assistance of isothermal titration calorimetry of model compounds, it was found that the changing ground-state distribution in differing solvent systems is driven entropically rather than enthalpically. Copyright © 2012 John Wiley & Sons, Ltd.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The solvent dependency of the ground-state distribution as well as the electrochemical switching behavior in a redox-active bistable donor–acceptor [2]catenane, containing bisthiotetrathiafulvalene (STTFS) and 1,5-dioxynaphthalene (DNP) recognition sites incorporated within a macrocyclic polyether encircled by the cyclobis(paraquat-p-phenylene) (CBPQT4+) ring, has been investigated. There are two translational isomers: (i) the ground-state co-conformation (GSCC) in which the CBPQT4+ ring encircles the STTFS unit and (ii) the metastable-state co-conformation (MSCC) in which the CBPQT4+ ring encircles the DNP unit. 1H NMR spectroscopy indicates that the ground-state distribution of GSCC to MSCC varies from approximately 1:1 in MeCN to 7:1 in MeCN : H2O (1:1, v/v) at 283 K. The reversible electrochemical switching behavior of the [2]catenane was confirmed by 1H NMR and UV−Vis spectroscopies, as well as by cyclic voltammetry (CV). Additionally, variable scan-rate CV studies were compared with simulated CV data and show that the ground-state distribution of GSCC to MSCC is about 30:1 in MeCN : H2O (1:1, v/v) at 298 K. With the assistance of isothermal titration calorimetry of model compounds, it was found that the changing ground-state distribution in differing solvent systems is driven entropically rather than enthalpically. Copyright © 2012 John Wiley & Sons, Ltd.
Fahrenbach, Albert C.; Zhu, Zhixue; Cao, Dennis; Liu, Wei-Guang; Li, Hao; Dey, Sanjeev K.; Basu, Subhadeep; Trabolsi, Ali; Botros, Youssry Y.; Goddard, William A.; Stoddart, J. Fraser
Radically Enhanced Molecular Switches Journal Article
In: J. Am. Chem. Soc., vol. 134, no. 39, pp. 16275−16288, 2012, ISSN: 0002-7863.
@article{fahrenbach_radically_2012,
title = {Radically Enhanced Molecular Switches},
author = {Fahrenbach, Albert C. and Zhu, Zhixue and Cao, Dennis and Liu, Wei-Guang and Li, Hao and Dey, Sanjeev K. and Basu, Subhadeep and Trabolsi, Ali and Botros, Youssry Y. and Goddard, William A. and Stoddart, J. Fraser},
url = {http://dx.doi.org/10.1021/ja306044r},
doi = {10.1021/ja306044r},
issn = {0002-7863},
year = {2012},
date = {2012-01-01},
urldate = {2013-05-17},
journal = {J. Am. Chem. Soc.},
volume = {134},
number = {39},
pages = {16275−16288},
abstract = {The mechanism governing the redox-stimulated switching behavior of a tristable [2]rotaxane consisting of a cyclobis(paraquat-p-phenylene) (CBPQT4+) ring encircling a dumbbell, containing tetrathiafulvalene (TTF) and 1,5-dioxynaphthalene (DNP) recognition units which are separated from each other along a polyether chain carrying 2,6-diisopropylphenyl stoppers by a 4,4?-bipyridinium (BIPY2+) unit, is described. The BIPY2+ unit acts to increase the lifetime of the metastable state coconformation (MSCC) significantly by restricting the shuttling motion of the CBPQT4+ ring to such an extent that the MSCC can be isolated in the solid state and is stable for weeks on end. As controls, the redox-induced mechanism of switching of two bistable [2]rotaxanes and one bistable [2]catenane composed of CBPQT4+ rings encircling dumbbells or macrocyclic polyethers, respectively, that contain a BIPY2+ unit with either a TTF or DNP unit, is investigated. Variable scan-rate cyclic voltammetry and digital simulations of the tristable and bistable [2]rotaxanes and [2]catenane reveal a mechanism which involves a bisradical state coconformation (BRCC) in which only one of the BIPY?+ units in the CBPQT2(?+) ring is oxidized to the BIPY2+ dication. This observation of the BRCC was further confirmed by theoretical calculations as well as by X-ray crystallography of the [2]catenane in its bisradical tetracationic redox state. It is evident that the incorporation of a kinetic barrier between the donor recognition units in the tristable [2]rotaxane can prolong the lifetime and stability of the MSCC, an observation which augurs well for the development of nonvolatile molecular flash memory devices.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The mechanism governing the redox-stimulated switching behavior of a tristable [2]rotaxane consisting of a cyclobis(paraquat-p-phenylene) (CBPQT4+) ring encircling a dumbbell, containing tetrathiafulvalene (TTF) and 1,5-dioxynaphthalene (DNP) recognition units which are separated from each other along a polyether chain carrying 2,6-diisopropylphenyl stoppers by a 4,4?-bipyridinium (BIPY2+) unit, is described. The BIPY2+ unit acts to increase the lifetime of the metastable state coconformation (MSCC) significantly by restricting the shuttling motion of the CBPQT4+ ring to such an extent that the MSCC can be isolated in the solid state and is stable for weeks on end. As controls, the redox-induced mechanism of switching of two bistable [2]rotaxanes and one bistable [2]catenane composed of CBPQT4+ rings encircling dumbbells or macrocyclic polyethers, respectively, that contain a BIPY2+ unit with either a TTF or DNP unit, is investigated. Variable scan-rate cyclic voltammetry and digital simulations of the tristable and bistable [2]rotaxanes and [2]catenane reveal a mechanism which involves a bisradical state coconformation (BRCC) in which only one of the BIPY?+ units in the CBPQT2(?+) ring is oxidized to the BIPY2+ dication. This observation of the BRCC was further confirmed by theoretical calculations as well as by X-ray crystallography of the [2]catenane in its bisradical tetracationic redox state. It is evident that the incorporation of a kinetic barrier between the donor recognition units in the tristable [2]rotaxane can prolong the lifetime and stability of the MSCC, an observation which augurs well for the development of nonvolatile molecular flash memory devices.
Forgan, Ross S.; Wang, Cheng; Friedman, Douglas C.; Spruell, Jason M.; Stern, Charlotte L.; Sarjeant, Amy A.; Cao, Dennis; Stoddart, J. Fraser
Donor–Acceptor Ring-in-Ring Complexes Journal Article
In: Chem. Eur. J., vol. 18, no. 1, pp. 202−212, 2012, ISSN: 1521-3765.
@article{forgan_donoracceptor_2012,
title = {Donor–Acceptor Ring-in-Ring Complexes},
author = {Forgan, Ross S. and Wang, Cheng and Friedman, Douglas C. and Spruell, Jason M. and Stern, Charlotte L. and Sarjeant, Amy A. and Cao, Dennis and Stoddart, J. Fraser},
url = {http://onlinelibrary.wiley.com/doi/10.1002/chem.201102919/abstract},
doi = {10.1002/chem.201102919},
issn = {1521-3765},
year = {2012},
date = {2012-01-01},
urldate = {2013-05-17},
journal = {Chem. Eur. J.},
volume = {18},
number = {1},
pages = {202−212},
abstract = {The self-assembly of three donor–acceptor ring-in-ring complexes, prepared from the π-electron-deficient tetracationic cyclophane, cyclobis(paraquat-4,4′-biphenylene), and three large π-electron-rich crown ethers (each 50-membered rings) containing dioxynaphthalene (DNP) and tetrathiafulvalene (TTF) units in pairs (DNP/DNP, DNP/TTF and TTF/TTF), is reported. 1H NMR spectroscopic analyses are indicative of the formation of 1:1 complexes in CD3CN, whilst the charge-transfer interactions between the DNP and TTF units of the crown ethers and the tetracationic cyclophane have permitted the measurement of binding constants of up to 4×103 M−1 in CH3CN to be made using UV/Vis spectroscopy. Ring-in-ring complexes are proposed as intermediates in the stepwise synthesis of molecular Borromean rings (BRs) comprised of three different rings. With the particular choice of crown ethers, the 1:1 complexes have polyether loops that protrude from the donor–acceptor recognition point above and below the mean plane of the tetracationic cyclophane, which, ideally, could conceivably bind dialkylammonium centers present in a third ring. X-ray crystallographic analyses of the solid-state superstructures of two of the three 1:1 complexes reveal, however, the presence of prodigious CH⋅⋅⋅O interactions between the polyether loops of the crown ethers and the rims of the cyclophane, no doubt stabilizing the complexes, but, at the same time, masking their potential recognition sites from further interactions that are essential to the subsequent emergence of the third ring. The solid-state superstructure of one of the crown ethers binding two dibenzylammonium ions provides some insight into the design requirements for the next generation of these systems; longer polyether loops may be required to allow optimal interactions between all components. It has become clear during a pursuit of the stepwise synthesis of the molecular BRs that, when designing complex mechanically interlocked molecules utilizing multiple recognition sites, the unsullied orthogonality of the recognition motifs is of the utmost importance.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The self-assembly of three donor–acceptor ring-in-ring complexes, prepared from the π-electron-deficient tetracationic cyclophane, cyclobis(paraquat-4,4′-biphenylene), and three large π-electron-rich crown ethers (each 50-membered rings) containing dioxynaphthalene (DNP) and tetrathiafulvalene (TTF) units in pairs (DNP/DNP, DNP/TTF and TTF/TTF), is reported. 1H NMR spectroscopic analyses are indicative of the formation of 1:1 complexes in CD3CN, whilst the charge-transfer interactions between the DNP and TTF units of the crown ethers and the tetracationic cyclophane have permitted the measurement of binding constants of up to 4×103 M−1 in CH3CN to be made using UV/Vis spectroscopy. Ring-in-ring complexes are proposed as intermediates in the stepwise synthesis of molecular Borromean rings (BRs) comprised of three different rings. With the particular choice of crown ethers, the 1:1 complexes have polyether loops that protrude from the donor–acceptor recognition point above and below the mean plane of the tetracationic cyclophane, which, ideally, could conceivably bind dialkylammonium centers present in a third ring. X-ray crystallographic analyses of the solid-state superstructures of two of the three 1:1 complexes reveal, however, the presence of prodigious CH⋅⋅⋅O interactions between the polyether loops of the crown ethers and the rims of the cyclophane, no doubt stabilizing the complexes, but, at the same time, masking their potential recognition sites from further interactions that are essential to the subsequent emergence of the third ring. The solid-state superstructure of one of the crown ethers binding two dibenzylammonium ions provides some insight into the design requirements for the next generation of these systems; longer polyether loops may be required to allow optimal interactions between all components. It has become clear during a pursuit of the stepwise synthesis of the molecular BRs that, when designing complex mechanically interlocked molecules utilizing multiple recognition sites, the unsullied orthogonality of the recognition motifs is of the utmost importance.
2011
Hanifi, David; Cao, Dennis; Klivansky, Liana M.; Liu, Yi
Novel C3-Symmetric n-Type Tris(aroyleneimidazole) and its Analogs: Synthesis, Physical Properties and Self-Assembly Journal Article
In: Chem. Commun., vol. 47, no. 12, pp. 3454−3456, 2011, ISSN: 1364-548X.
@article{hanifi_novel_2011,
title = {Novel C3-Symmetric n-Type Tris(aroyleneimidazole) and its Analogs: Synthesis, Physical Properties and Self-Assembly},
author = {Hanifi, David and Cao, Dennis and Klivansky, Liana M. and Liu, Yi},
url = {http://pubs.rsc.org/en/content/articlelanding/2011/cc/c0cc04753h},
doi = {10.1039/C0CC04753H},
issn = {1364-548X},
year = {2011},
date = {2011-01-01},
urldate = {2013-05-17},
journal = {Chem. Commun.},
volume = {47},
number = {12},
pages = {3454−3456},
abstract = {Novel n-type C3-symmetric materials are synthesized and shown to have desirable bandgap, broad absorption and high thermal stability, thus pose as viable candidates for organic photovoltaics. The strong intermolecular interactions among the extended π-surfaces beget the self-assembly of nanofibers.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Novel n-type C3-symmetric materials are synthesized and shown to have desirable bandgap, broad absorption and high thermal stability, thus pose as viable candidates for organic photovoltaics. The strong intermolecular interactions among the extended π-surfaces beget the self-assembly of nanofibers.
2010
Koshkakaryan, Gayane; Jiang, Peng; Altoe, Virginia; Cao, Dennis; Klivansky, Liana M.; Zhang, Yue; Chung, Sungwook; Katan, Allard; Martin, Florent; Salmeron, Miquel; Ma, Biwu; Aloni, Shaul; Liu, Yi
Multilayered Nanofibers from Stacks of Single-Molecular Thick Nanosheets of Hexakis(alkoxy)triphenylenes Journal Article
In: Chem. Commun., vol. 46, no. 45, pp. 8579−8581, 2010, ISSN: 1364-548X.
@article{koshkakaryan_multilayered_2010,
title = {Multilayered Nanofibers from Stacks of Single-Molecular Thick Nanosheets of Hexakis(alkoxy)triphenylenes},
author = {Koshkakaryan, Gayane and Jiang, Peng and Altoe, Virginia and Cao, Dennis and Klivansky, Liana M. and Zhang, Yue and Chung, Sungwook and Katan, Allard and Martin, Florent and Salmeron, Miquel and Ma, Biwu and Aloni, Shaul and Liu, Yi},
url = {http://pubs.rsc.org/en/content/articlelanding/2010/cc/c0cc03942j},
doi = {10.1039/C0CC03942J},
issn = {1364-548X},
year = {2010},
date = {2010-11-01},
urldate = {2013-05-17},
journal = {Chem. Commun.},
volume = {46},
number = {45},
pages = {8579−8581},
abstract = {Symmetrically substituted hexakis(alkoxy)triphenylene (HAT) derivatives were assembled into single molecular thick 2D nanosheets, which stacked further to give multilayered nanofibers through a convenient solution process. Detailed information on molecular arrangement was unraveled by various imaging techniq},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Symmetrically substituted hexakis(alkoxy)triphenylene (HAT) derivatives were assembled into single molecular thick 2D nanosheets, which stacked further to give multilayered nanofibers through a convenient solution process. Detailed information on molecular arrangement was unraveled by various imaging techniq
Cao, Dennis; Amelia, Matteo; Klivansky, Liana M.; Koshkakaryan, Gayane; Khan, Saeed I.; Semeraro, Monica; Silvi, Serena; Venturi, Margherita; Credi, Alberto; Liu, Yi
Probing Donor−Acceptor Interactions and Co-Conformational Changes in Redox Active Desymmetrized [2]Catenanes Journal Article
In: J. Am. Chem. Soc., vol. 132, no. 3, pp. 1110−1122, 2010, ISSN: 0002-7863.
@article{cao_probing_2010,
title = {Probing Donor−Acceptor Interactions and Co-Conformational Changes in Redox Active Desymmetrized [2]Catenanes},
author = {Cao, Dennis and Amelia, Matteo and Klivansky, Liana M. and Koshkakaryan, Gayane and Khan, Saeed I. and Semeraro, Monica and Silvi, Serena and Venturi, Margherita and Credi, Alberto and Liu, Yi},
url = {http://dx.doi.org/10.1021/ja909041g},
doi = {10.1021/ja909041g},
issn = {0002-7863},
year = {2010},
date = {2010-01-01},
urldate = {2013-05-17},
journal = {J. Am. Chem. Soc.},
volume = {132},
number = {3},
pages = {1110−1122},
abstract = {We describe the synthesis and characterization of a series of desymmetrized donor?acceptor [2]catenanes where different donor and acceptor units are assembled within a confined catenated geometry. Remarkable translational selectivity is maintained in all cases, including two fully desymmetrized [2]catenanes where both donors and acceptors are different, as revealed by X-ray crystallography in the solid state, and by 1H NMR spectroscopy and electrochemistry in solution. In all desymmetrized [2]catenanes the co-conformation is dominated by the strongest donor and acceptor pairs, whose charge-transfer interactions also determine the visible absorption properties. Voltammetric and spectroelectrochemical experiments show that the catenanes can be reversibly switched among as many as seven states, characterized by distinct electronic and optical properties, by electrochemical stimulation in a relatively narrow and easily accessible potential window. Moreover in some of these compounds the oxidation of the electron donor units or the reduction of the electron acceptor ones causes the circumrotation of one molecular ring with respect to the other. These features make these compounds appealing for the development of molecular electronic devices and mechanical machines.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
We describe the synthesis and characterization of a series of desymmetrized donor?acceptor [2]catenanes where different donor and acceptor units are assembled within a confined catenated geometry. Remarkable translational selectivity is maintained in all cases, including two fully desymmetrized [2]catenanes where both donors and acceptors are different, as revealed by X-ray crystallography in the solid state, and by 1H NMR spectroscopy and electrochemistry in solution. In all desymmetrized [2]catenanes the co-conformation is dominated by the strongest donor and acceptor pairs, whose charge-transfer interactions also determine the visible absorption properties. Voltammetric and spectroelectrochemical experiments show that the catenanes can be reversibly switched among as many as seven states, characterized by distinct electronic and optical properties, by electrochemical stimulation in a relatively narrow and easily accessible potential window. Moreover in some of these compounds the oxidation of the electron donor units or the reduction of the electron acceptor ones causes the circumrotation of one molecular ring with respect to the other. These features make these compounds appealing for the development of molecular electronic devices and mechanical machines.
Koshkakaryan, Gayane; Cao, Dennis; Klivansky, Liana M.; Teat, Simon J.; Tran, Jasper L.; Liu, Yi
Dual Selectivity Expressed in [2 + 2 + 1] Dynamic Clipping of Unsymmetrical [2]Catenanes Journal Article
In: Org. Lett., vol. 12, no. 7, pp. 1528−1531, 2010, ISSN: 1523-7060.
@article{koshkakaryan_dual_2010,
title = {Dual Selectivity Expressed in [2 + 2 + 1] Dynamic Clipping of Unsymmetrical [2]Catenanes},
author = {Koshkakaryan, Gayane and Cao, Dennis and Klivansky, Liana M. and Teat, Simon J. and Tran, Jasper L. and Liu, Yi},
url = {http://dx.doi.org/10.1021/ol100215c},
doi = {10.1021/ol100215c},
issn = {1523-7060},
year = {2010},
date = {2010-01-01},
urldate = {2013-05-17},
journal = {Org. Lett.},
volume = {12},
number = {7},
pages = {1528−1531},
abstract = {A π-templated dynamic [2 + 2 + 1] clipping protocol is established for the synthesis of [2]catenanes from two parts dialdehyde, two parts diamine, and one part tetracationic cyclophane. It is further diversified for the selective formation of an unsymmetrical [2]catenane showing great translational selectivity by employing two different dialdehydes in a one-pot reaction. The dual selectivity and the dynamic nature are verified by 1H NMR spectroscopy, X-ray single-crystal structural studies, and exchange experiments.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
A π-templated dynamic [2 + 2 + 1] clipping protocol is established for the synthesis of [2]catenanes from two parts dialdehyde, two parts diamine, and one part tetracationic cyclophane. It is further diversified for the selective formation of an unsymmetrical [2]catenane showing great translational selectivity by employing two different dialdehydes in a one-pot reaction. The dual selectivity and the dynamic nature are verified by 1H NMR spectroscopy, X-ray single-crystal structural studies, and exchange experiments.
2009
Koshkakaryan, Gayane; Klivansky, Liana M.; Cao, Dennis; Snauko, Marian; Teat, Simon J.; Struppe, Jochem O.; Liu, Yi
In: J. Am. Chem. Soc., vol. 131, no. 6, pp. 2078−2079, 2009, ISSN: 0002-7863.
@article{koshkakaryan_alternative_2009,
title = {Alternative Donor−Acceptor Stacks from Crown Ethers and Naphthalene Diimide Derivatives: Rapid, Selective Formation from Solution and Solid State Grinding},
author = {Koshkakaryan, Gayane and Klivansky, Liana M. and Cao, Dennis and Snauko, Marian and Teat, Simon J. and Struppe, Jochem O. and Liu, Yi},
url = {http://dx.doi.org/10.1021/ja809088v},
doi = {10.1021/ja809088v},
issn = {0002-7863},
year = {2009},
date = {2009-01-01},
urldate = {2013-05-17},
journal = {J. Am. Chem. Soc.},
volume = {131},
number = {6},
pages = {2078−2079},
abstract = {The atypical 1:2 complexation between an electron-rich crown ether host and electron-deficient naphthalene diimide-based guests led to the formation of alternative donor?acceptor (ADA) stacks. The ADA stacks can be expediently obtained in high yield as polycrystalline aggregates from solution. More remarkably, the high degree of organization has also been realized in a simple solid-to-solid mechanical grinding process. The solid-state structures have been verified by solid-state NMR spectroscopy, single crystal, and powder X-ray diffraction analysis. The current findings not only provide convenient ways of obtaining novel donor?acceptor stacks involving a macrocyclic host but also represent an important step in transferring electroactive host?guest systems from solution to the solid state.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
The atypical 1:2 complexation between an electron-rich crown ether host and electron-deficient naphthalene diimide-based guests led to the formation of alternative donor?acceptor (ADA) stacks. The ADA stacks can be expediently obtained in high yield as polycrystalline aggregates from solution. More remarkably, the high degree of organization has also been realized in a simple solid-to-solid mechanical grinding process. The solid-state structures have been verified by solid-state NMR spectroscopy, single crystal, and powder X-ray diffraction analysis. The current findings not only provide convenient ways of obtaining novel donor?acceptor stacks involving a macrocyclic host but also represent an important step in transferring electroactive host?guest systems from solution to the solid state.
Klivansky, Liana M.; Koshkakaryan, Gayane; Cao, Dennis; Liu, Yi
Linear π-Acceptor-Templated Dynamic Clipping to Macrobicycles and [2]Rotaxanes Journal Article
In: Angew. Chem. Int. Ed., vol. 48, no. 23, pp. 4185−4189, 2009, ISSN: 1521-3773.
@article{klivansky_linear_2009,
title = {Linear π-Acceptor-Templated Dynamic Clipping to Macrobicycles and [2]Rotaxanes},
author = {Klivansky, Liana M. and Koshkakaryan, Gayane and Cao, Dennis and Liu, Yi},
url = {http://onlinelibrary.wiley.com/doi/10.1002/anie.200900716/abstract},
doi = {10.1002/anie.200900716},
issn = {1521-3773},
year = {2009},
date = {2009-01-01},
urldate = {2013-05-17},
journal = {Angew. Chem. Int. Ed.},
volume = {48},
number = {23},
pages = {4185−4189},
abstract = {Cage me! A linear dumbbell-shaped bipyridinium molecule can template cage formation around itself through sixfold imine bond formation to give an interlocked [2]rotaxane as the single product (see picture). This highly efficient [2+3] clipping occurs despite the symmetry mismatch between the template and the formed macrobicycle.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Cage me! A linear dumbbell-shaped bipyridinium molecule can template cage formation around itself through sixfold imine bond formation to give an interlocked [2]rotaxane as the single product (see picture). This highly efficient [2+3] clipping occurs despite the symmetry mismatch between the template and the formed macrobicycle.